
Семейна хиперхолестеролемия: какво е и как да се лекува
Фамилна хиперхолестеролемия, рядко генетично заболяване, което причинява сериозни последици за здравето (напр. Инфаркт и инсулт), които изискват подходящи мерки за ограничаване с подходящи лекарства и подходящ начин на живот.
Какво означава хиперхолестеролемия? И какво означава фамилна?
Хиперхолестеролемията е клинично състояние, характеризиращо се с прекомерен холестерол в кръвта.
Холестеролът е компонент на липидите, който обикновено се приема с диетата, но който може да се произвежда и от организма.
Холестеролът е много важен за живота, защото се използва за образуване на клетъчни мембрани (за да се осигури тяхната функция), предпазва невроните и черепните нерви.
Холестеролът се използва и за синтеза на други молекули, три от които са жлъчни киселини (важни за храносмилането), някои хормони и витамин D.
Само ако е налице над тези изисквания, това може да причини сериозни щети.
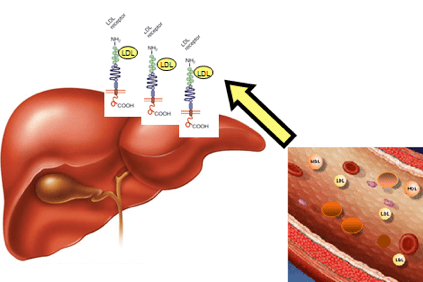
В случай на хиперхолестеролемия, холестеролът се натрупва в кръвта под формата на леки липопротеини (LDL), определени агрегати от мазнини и протеини, наричани още „лош холестерол“, които подпомагат образуването на плаки в артериалната стена (атеросклеротични плаки).
Това се случва, когато пациентите имат толкова високи нива на холестерол, че не могат да го елиминират чрез физиологичните механизми на черния дроб.
Натрупването на LDL в кръвта в крайна сметка води до образуването на атерома (физическо препятствие за нормалния кръвен поток), което може да доведе до по-сериозни последици като ангина, инфаркт, инсулт, но също така и в други органи като мозъка, бъбреците, белите дробове и самия черен дроб.
Хиперхолестеролемията е позната, когато се предава на потомство
Това се дължи на модификации на ген, който съдържа информацията за образуване на чернодробен протеин, LDLR (LDLR) рецептор, който разпознава LDL, премахва го от кръвния поток и го транспортира до чернодробните клетки, които след това го отстраняват. Тази генна промяна води до натрупване на LDL в кръвта.
Към днешна дата има повече от 600 известни промени в LDL гена, които причиняват фамилна хиперхолестеролемия.
Също така, хората с това разстройство могат да внесат промени в други гени, участващи в метаболизма на липопротеините като APOB гена (апопротеин В в LDL) и гена PCSK9 (протеин, който разгражда LDL рецепторите).
Въпреки че е известно, че натрупването на холестерол може да бъде вредно, се изчислява, че до една трета от инфарктите, настъпили преди 40-годишна възраст, се дължат на недиагностицирана или неправилно лекувана семейна хиперхолестеролемия.
По-често срещана е многофакторната хиперхолестеролемия, причинена от фактори на околната среда (диета с високо съдържание на мазнини, особено ако е свързана с физическо бездействие), дори при наличие на предразполагащи генетични фактори.
Формите, в които се проявява семейната хиперхолестеролемия
Това заболяване може да се прояви в 2 различни форми: по-малко сериозна (хетерозиготна, 1 случай на 500 индивида) и по-сериозна (хомозиготна, 1 случай на 1,000,000 1,2 XNUMX индивида) XNUMX.
Хетерозиготната форма често протича безсимптомно и се диагностицира само въз основа на нивата на холестерола в кръвта.
Черният дроб се бори да елиминира LDL, тъй като LDL рецепторите се произвеждат в недостатъчен брой, което води до 2 или 3-кратно увеличение на кръвните нива в сравнение с нормалните стойности.
Тази форма може да доведе до повишен риск от сърдечно-съдови заболявания в зряла възраст.
Хомозиготната форма се характеризира с появата на сърдечно-съдови заболявания дори в млада възраст и наличието на характерни натрупвания на мазнини като ксантоми (жълтеникави възли по кокалчетата на ръцете и ахилесовото сухожилие) и ксантелазма (жълтеникави плочи по клепачите и около очите).
Генетичният дефект се наследява и от двамата родители и рискът от инфаркт при липса на терапия вече се наблюдава около 15-20 годишна възраст.
Всъщност при това състояние черният дроб не успява да метаболизира липопротеините, които остават в кръвта, които след това се натрупват, което води до описаните по-горе дисфункции и създава ситуация, несъвместима с живота.
Какви са шансовете за предаване на болестта на деца?
Тъй като всеки от нас има по две копия на всеки ген, всяко наследено от родител, ние ще имаме хетерозиготна форма, когато наследяваме едно изменено и едно здраво копие.
Напротив, ще имаме хомозиготна форма, ако наследим и двете копия на болния ген от двамата родители. Трябва да се каже, че тази форма е много рядка, защото се появява само когато и двамата родители имат гена, който причинява заболяването, и следователно всеки от тях може да предаде копие на една и съща генна промяна.
Фамилната хиперхолестеролемия се нарича хетерозиготна патология, съставена в редките случаи, при които два различни вида генни промени се наследяват, по един от всеки родител.
Децата с риск от развитие на болестта трябва да бъдат диагностицирани и лекувани по-рано. Днес е възможно да се диагностицира болестта чрез генетични тестове, които търсят грешки (генни промени) на LDLR, ApoB и PCSK9 гени.
Следователно само формите на семейната хиперхолестеролемия са трансмисивни.
Многофакторните форми, които се появяват главно поради неправилен начин на живот (натрупване на холестерол с диетата), също могат да се характеризират с генетично предразположение.
Всъщност тези субекти могат да имат генетичен дефицит, който компрометира способността на организма да компенсира адекватно излишните липиди в диетата.
Например, когато черният дроб е наситен с холестерол, поет с храна, производството на рецептори, които улавят LDL, циркулиращи в кръвта, може да бъде потиснато.
Ситуацията е много подобна на тази, която се случва по време на семейната хиперхолестеролемия, макар и по-малко тежка.
В този случай концентрацията на общ холестерол в кръвта е над нормата и обикновено е между 240 и 350 mg / dl в сравнение с нормалната, която обикновено е определена около 200 - 240 mg / dl.

Любопитство: тази болест може да е била предавана през вековете
Внимателното наблюдение на известната картина на Мона Лиза показва възможността Мона Лиза още в млада възраст да страда от семейна хиперхолестеролемия.
Всъщност Леонардо също вярно изобразява типичните мастни натрупвания по ръцете и в близост до очите (сега известни като ксантоми и ксантелазма), които със сигурност показват наличието на патологията.
Ако това е вярно, се показва, че семейната хиперхолестеролемия е болест, запазена през вековете (случай, документиран още през 1500 г.).
Какви са възможностите за лечение на това заболяване днес?
Възможността за излекуване на заболяването зависи от неговата тежест. Също така рисковите фактори (диета, тютюнопушене, възраст, семейство и лична история на хиперхолестеролемия, наличие на други заболявания) могат да влошат общата картина на заболяването.
За лечение на семейната хиперхолестеролемия могат да се използват лекарства от ново поколение, но в същото време е необходимо да се действа с подходяща модификация на начина на живот.
В случаите на средна тежест (хетерозиготна фамилна хиперхолестеролемия), лекарствена терапия, базирана на статини (инхибитори на синтеза на холестерол, които индуцират повишен синтез на LDLR рецептор) и комбинацията с езетимиб (инхибитори на абсорбцията на холестерола) или PCSK9 инхибитори (т.е. унищожават чернодробните рецептори, които улавят LDLR) подобрява активността на здравословното копие на LDLR гена и намалява натрупването на холестерол в кръвта.
Наскоро одобрена е и бемпедоева киселина, лекарство, което действа в черния дроб чрез инхибиране на ензима АТР цитратна лиаза, молекула, участваща в процеса на ендогенен синтез на холестерол.
Този механизъм на действие ни позволява да действаме върху количеството произведен холестерол, действайки нагоре от мястото на действие на статините, и да стимулира експресията на LDL рецептори, за да компенсира намаления синтез.
За разлика от статините, бемпедоевата киселина не е активна в скелетните мускули, което намалява възможността от нежелани събития, типични за статините.
Това лекарство може също да бъде свързано с езетимиб с благоприятни ефекти.
Хомозиготната фамилна хиперхолестеролемия доскоро се смяташе за нелечимо заболяване.
Терапията със статини не е ефективна при тази патология. Всъщност статините, които действат върху механизмите, които водят до производството на ендогенен холестерол, не могат да стимулират синтеза на LDL рецептори.
За да се елиминира LDL холестерола от тялото на тези пациенти, плазмафереза, техника, която позволява филтриране на кръвта чрез елиминиране на мазнини, подобно на това, което се прави с диализа, когато бъбреците не работят.
Това обаче е инвазивна процедура с отрицателно въздействие върху качеството на живот на пациентите.
Най-новите изследвания върху семейната хиперхолестеролемия
През последните години изследванията доведоха до разработването на специфични лекарства и за хомозиготната форма на фамилна хиперхолестеролемия, значително подобрявайки очакванията и качеството на живот на пациентите, които страдат от нея.
Например, лекарството ломитапид (приемано през устата) води до значително по-ниски плазмени нива на LDL-холестерол при тези пациенти.
Ломитапид инхибира микрозомален триглицериден транспортен протеин (MTP), който позволява включването на тези липиди заедно с апопротеина В100 в зараждащи се VLDL.
В резултат на това, апопротеинът B100 и LDL са намалени дори при пациенти, при които заболяването е причинило пълното отсъствие на LDL рецептора.
Друго лекарство от ново поколение е mipomersen, антисенс олигонуклеотид, способен да разгражда B100 апопротеините, които участват в образуването на LDL, като по този начин намалява броя на последните.
Еволокумаб и алирокумаб са две моноклонални антитела, които инхибират активността на PCSK9 протеина в кръвта.
За да влязат в сила обаче тези две лекарства, трябва да присъстват и да функционират поне малка част от LDL рецепторите.
Сред лекарствата, които все още са в клинично развитие, е включването на биологично лекарство (т.нар. SiRNA), което блокира ДНК транскрипцията и синтеза на белтъка PCSK9 в черния дроб.
Наскоро бяха постигнати положителни резултати с евинакумаб, инхибитор на моноклонално антитяло на протеина подобен на ангиопоетин 3 (ANGPTL3), молекула, синтезирана от черния дроб, чиято роля е да повишава нивата на холестерола-LLDL и триглицеридите, предотвратявайки тяхното разграждане.
Това ново лекарство също показва ефективност при пациенти, при които LDL рецепторът не функционира3,4.
Наскоро бяха направени и някои опити за разбиране дали генната терапия с въвеждането на здравия ген в ДНК на пациента може да работи5.
От този списък с възможни терапии за фамилна хиперхолестеролемия е възможно да се разбере колко изследвания се правят, за да се осигурят решения за тези пациенти, които, макар и рядко, са засегнати от това сериозно заболяване, намалявайки риска от фатални последици.
Както винаги се случва с новите лекарства, добре е да се използва предпазливост при тяхната хронична употреба, тъй като знанията за възможните им странични ефекти ще бъдат по-ясни само с опит (фармакологична бдителност), така че тяхната употреба в педиатрията трябва да бъде внимателно оценена, разбира се с крайната цел да предложи на тези „малки“ пациенти възможността да водят „нормален“ живот.
Библиографски и ситографски справки за статията за семейната хиперхолестеролемия
1 2019 ESC / EAS насоки за лечение на дислипидемии: Липидна модификация за намаляване на сърдечно-съдовия риск. Автори / членове на работната група; Комитет за практически насоки на ESC (CPG); ESC Национални сърдечни дружества. Атеросклероза. 2019 ноември; 290: 140-205. doi: 10.1016 / j. атеросклероза.2019.08.014.
2 Редки дислипидемии, от фенотип до генотип до управление: консенсус на Европейската група за атеросклероза. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Ланцетен диабет Ендокринол. 2020 г. януари; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Липопротеин (а) Понижаване - от липопротеинова афереза към антисенс олигонуклеотиден подход. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 г. 3 юли; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 Терапия за понижаване на LDL-холестерола. Pirillo A, Norata GD, Catapano AL.Handb Exp Pharmacol. 2020 г. 30 април. Doi: 10.1007 / 164_2020_361.
5 Преглед на генетични и клетъчни терапии за фамилна хиперхолестеролемия. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A.Pharmacol Res. 2019 май; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.
Прочетете още:
Прочетете статията на италиански