
Hipercolesterolemia familiar: que es y como tratarla
Hipercolesterolemia familiar, una enfermedad genética rara que causa graves consecuencias para la salud (por ejemplo, ataque cardíaco y accidente cerebrovascular) que requiere medidas de contención adecuadas con medicación adecuada y un estilo de vida adecuado.
¿Qué significa hipercolesterolemia? ¿Y qué significa familiar?
La hipercolesterolemia es una condición clínica caracterizada por un exceso de colesterol en la sangre.
El colesterol es un componente de los lípidos, que generalmente se toma con la dieta, pero que también puede ser producido por el cuerpo.
El colesterol es muy importante para la vida porque se utiliza para la formación de membranas celulares (para asegurar su función), protege las neuronas y los nervios craneales.
El colesterol también se usa para la síntesis de otras moléculas, tres de las cuales son ácidos biliares (importantes para la digestión), algunas hormonas y vitamina D.
Solo si está presente por encima de estos requisitos puede causar daños graves.
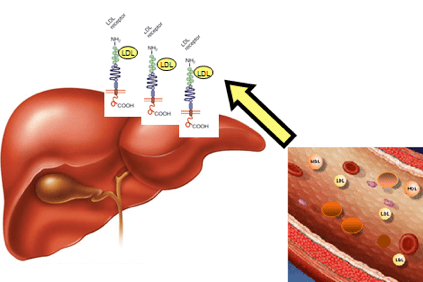
En el caso de la hipercolesterolemia, el colesterol se acumula en la sangre en forma de lipoproteínas ligeras (LDL), agregados particulares de grasas y proteínas, también llamados “colesterol malo” que favorecen la formación de placas en la pared arterial (placas ateroscleróticas).
Esto sucede cuando los pacientes tienen niveles de colesterol tan altos que no pueden eliminarlo a través de los mecanismos fisiológicos del hígado.
La acumulación de LDL en la sangre eventualmente conduce a la formación del ateroma (una obstrucción física del flujo sanguíneo normal) que puede tener consecuencias más graves como angina, ataque cardíaco, accidente cerebrovascular, pero también en otros órganos como el cerebro. riñones, pulmones y el propio hígado.
La hipercolesterolemia es familiar cuando se transmite a la descendencia.
Esto se debe a modificaciones de un gen que contiene la información para producir una proteína hepática, el receptor LDLR (LDLR), que reconoce el LDL, lo elimina del torrente sanguíneo y lo transporta a las células del hígado, que luego lo eliminan. Esta alteración genética provoca la acumulación de LDL en la sangre.
Hasta la fecha, existen más de 600 alteraciones conocidas en el gen LDL que causan hipercolesterolemia familiar.
Además, las personas con este trastorno pueden traer cambios en otros genes involucrados en el metabolismo de las lipoproteínas como el gen APOB (apoproteína B en LDL) y el gen PCSK9 (una proteína que degrada los receptores de LDL).
Aunque se sabe que la acumulación de colesterol puede ser dañina, se estima que hasta un tercio de los ataques cardíacos que ocurren antes de los 40 años se deben a una hipercolesterolemia familiar no diagnosticada o tratada de manera inapropiada.
Más común es la hipercolesterolemia multifactorial causada por factores ambientales (una dieta rica en grasas, especialmente si se asocia con inactividad física) incluso en presencia de factores genéticos predisponentes.
Las formas en las que se manifiesta la hipercolesterolemia familiar
Esta enfermedad puede presentarse de 2 formas diferentes: una menos grave (heterocigótica, 1 caso cada 500 individuos) y una más grave (homocigótica, 1 caso cada 1,000,000 de individuos) 1,2.
La forma heterocigótica a menudo es asintomática y se diagnostica solo en función de los niveles de colesterol en sangre.
El hígado lucha por eliminar el LDL porque los receptores de LDL se producen en cantidades insuficientes, lo que resulta en un aumento de 2 o 3 veces en los niveles sanguíneos en comparación con los valores normales.
Esta forma puede conducir a un mayor riesgo de enfermedad cardiovascular en la edad adulta.
La forma homocigótica se caracteriza por la aparición de enfermedades cardiovasculares incluso a una edad temprana y la presencia de acumulaciones características de grasa como xantomas (nódulos amarillentos en los nudillos de las manos y el tendón de Aquiles) y xantelasma (placas amarillentas en los párpados y alrededor los ojos).
El defecto genético se hereda de ambos padres y ya se observa el riesgo de infarto en ausencia de terapia alrededor de los 15-20 años.
De hecho, en esta condición, el hígado no logra metabolizar las lipoproteínas que quedan en la sangre, que luego se acumulan dando lugar a las disfunciones descritas anteriormente y crean una situación incompatible con la vida.
¿Cuáles son las posibilidades de transmisión de la enfermedad a los niños?
Dado que cada uno de nosotros tiene dos copias de cada gen, cada una heredada de un padre, tendremos la forma heterocigota cuando heredemos una copia alterada y otra sana.
Por el contrario, tendremos la forma homocigótica si heredamos ambas copias del gen enfermo de ambos padres. Hay que decir que esta forma es muy rara porque ocurre solo cuando ambos padres tienen el gen que causa la enfermedad y por lo tanto cada uno de ellos puede transmitir una copia del mismo gen alterado.
La hipercolesterolemia familiar se denomina patología heterocigótica compuesta en los raros casos en los que se heredan dos tipos diferentes de alteraciones genéticas, una de cada progenitor.
Los niños en riesgo de desarrollar la enfermedad deben ser diagnosticados y tratados temprano. Hoy en día es posible diagnosticar la enfermedad mediante pruebas genéticas que buscan errores (alteraciones genéticas) en los genes LDLR, ApoB y PCSK9.
Por tanto, solo las formas de hipercolesterolemia familiar son transmisibles.
Sin embargo, las formas multifactoriales que ocurren principalmente por un estilo de vida incorrecto (acumulación de colesterol con la dieta), también pueden caracterizarse por una predisposición genética.
De hecho, estos sujetos pueden presentar déficits genéticos que comprometen la capacidad del organismo para compensar adecuadamente el exceso de lípidos en la dieta.
Por ejemplo, cuando el hígado está saturado de colesterol que se ingiere con los alimentos, se puede suprimir la producción de receptores que capturan el LDL que circula en la sangre.
Una situación muy parecida a la que se da durante la hipercolesterolemia familiar aunque menos grave.
En este caso, la concentración de colesterol total en sangre está por encima de lo normal y suele estar entre 240 y 350 mg / dl en comparación con la normal establecida convencionalmente alrededor de 200-240 mg / dl.

Una curiosidad: esta enfermedad puede haberse transmitido a lo largo de los siglos
Una observación cuidadosa del famoso cuadro de la Mona Lisa muestra la posibilidad de que Mona Lisa ya a una edad temprana padeciera hipercolesterolemia familiar.
De hecho, Leonardo también representa fielmente los típicos depósitos de grasa en las manos y cerca de los ojos (ahora conocidos como xantomas y xantelasma) que indican con certeza la presencia de la patología.
De ser así, se demuestra que la hipercolesterolemia familiar es una enfermedad conservada a lo largo de los siglos (caso documentado ya en 1500).
¿Cuáles son las posibilidades de tratar esta enfermedad hoy?
La posibilidad de curar la enfermedad depende de su gravedad. Además, los factores de riesgo (dieta, tabaquismo, edad, antecedentes familiares y personales de hipercolesterolemia, presencia de otras enfermedades) pueden empeorar el cuadro general de la enfermedad.
Para tratar la hipercolesterolemia familiar se pueden utilizar fármacos de nueva generación, pero al mismo tiempo, es necesario actuar con una modificación adecuada del estilo de vida.
En casos de gravedad intermedia (hipercolesterolemia familiar heterocigótica), la terapia con medicamentos a base de estatinas (inhibidores de la síntesis de colesterol que inducen un aumento de la síntesis del receptor de LDLR) y la combinación con ezetimiba (inhibidores de la absorción de colesterol) o fármacos inhibidores de PCSK9 (es decir, inhibidores de la proteína PCSK9 cuya función destruir los receptores hepáticos que capturan LDLR) mejora la actividad de la copia sana del gen LDLR y reduce la acumulación de colesterol en la sangre.
Recientemente aprobado también está el ácido bempedoico, un fármaco que actúa en el hígado inhibiendo la enzima ATP citrato liasa, una molécula implicada en el proceso de síntesis endógena del colesterol.
Este mecanismo de acción nos permite actuar sobre la cantidad de colesterol producido, actuando corriente arriba del sitio de acción de las estatinas, y estimular la expresión de los receptores de LDL para compensar la síntesis reducida.
A diferencia de las estatinas, el ácido bempedoico no es activo en el músculo esquelético, lo que disminuye la posibilidad de tener eventos no deseados típicos de las estatinas.
Este fármaco también puede asociarse con ezetimiba con efectos favorables.
La hipercolesterolemia familiar homocigótica se consideraba hasta hace poco una enfermedad incurable.
La terapia a base de estatinas no es eficaz en esta patología. De hecho, las estatinas que actúan sobre los mecanismos que conducen a la producción de colesterol endógeno no pueden estimular la síntesis de los receptores de LDL.
Para eliminar el colesterol LDL del organismo de estos pacientes se realiza la plasmaféresis, técnica que permite filtrar la sangre eliminando grasas, de forma similar a lo que se hace con la diálisis cuando los riñones no funcionan.
Sin embargo, este es un procedimiento invasivo con un impacto negativo en la calidad de vida de los pacientes.
Las últimas investigaciones sobre hipercolesterolemia familiar
En los últimos años, la investigación ha llevado al desarrollo de fármacos específicos también para la forma homocigótica de hipercolesterolemia familiar, mejorando enormemente la expectativa y calidad de vida de los pacientes que la padecen.
Por ejemplo, el fármaco lomitapida (tomado por vía oral) conduce a niveles de colesterol LDL en plasma notablemente más bajos en estos pacientes.
La lomitapida inhibe una proteína transportadora de triglicéridos microsomal (MTP) que permite la incorporación de estos lípidos junto con la apoproteína B100 en VLDL nacientes.
Como resultado, la apoproteína B100 y LDL se reducen incluso en pacientes en los que la enfermedad ha causado la ausencia total del receptor de LDL.
Otro fármaco de nueva generación es el mipomersen, un oligonucleótido antisentido capaz de degradar las apoproteínas B100 que intervienen en la formación de LDL, reduciendo así el número de estas últimas.
Evolocumab y alirocumab son dos anticuerpos monoclonales que inhiben la actividad de la proteína PCSK9 en la sangre.
Sin embargo, para que estos dos fármacos surtan efecto, al menos una pequeña parte de los receptores de LDL debe estar presente y funcionando.
Entre los fármacos que aún se encuentran en desarrollo clínico se encuentra la inclusión de un fármaco biológico (el denominado ARNip) que bloquea la transcripción del ADN y la síntesis de la proteína PCSK9 en el hígado.
Recientemente se han obtenido resultados positivos con evinacumab, un anticuerpo monoclonal inhibidor de la proteína similar angiopoyetina 3 (ANGPTL3), una molécula sintetizada por el hígado cuya función es aumentar los niveles de colesterol-LLDL y triglicéridos evitando su degradación.
Este nuevo fármaco también ha mostrado eficacia en pacientes en los que el receptor de LDL no funciona3,4.
Recientemente también se han realizado algunos intentos para comprender si la terapia génica, con la introducción del gen sano en el ADN del paciente, podría funcionar5.
A partir de esta lista de posibles terapias para la hipercolesterolemia familiar, es posible comprender cuánta investigación se está realizando para brindar soluciones a estos pacientes que, aunque raros, se ven afectados por esta grave enfermedad, reduciendo el riesgo de consecuencias fatales.
Sin embargo, como siempre ocurre con los medicamentos nuevos, es bueno tener una palabra de precaución sobre su uso crónico ya que el conocimiento sobre sus posibles efectos secundarios será más claro solo con la experiencia (farmacovigilancia), por lo que su uso en pediatría debe evaluarse cuidadosamente. por supuesto con el fin último de ofrecer a estos “pequeños” pacientes la posibilidad de llevar una vida “normal”.
Referencias bibliográficas y sitográficas del artículo sobre hipercolesterolemia familiar
1 Guía ESC / EAS 2019 para el manejo de dislipidemias: modificación de lípidos para reducir el riesgo cardiovascular. Autores / miembros del grupo de trabajo; Comité de Guías Prácticas (CPG) de la ESC; ESC Sociedades Nacionales de Cardiología. Aterosclerosis. Noviembre de 2019; 290: 140-205. doi: 10.1016 / j. ateroesclerosis.2019.08.014.
2 Dislipidemias raras, del fenotipo al genotipo y al tratamiento: declaración de consenso del grupo de trabajo de la Sociedad Europea de Aterosclerosis. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet Diabetes Endocrinol. Enero de 2020; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoproteína (a) Reducción de aféresis de lipoproteínas a enfoque de oligonucleótidos antisentido. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 3 de julio; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 Terapia para reducir el colesterol LDL. Pirillo A, Norata GD, Catapano AL. Handb Exp Pharmacol. 2020 30 de abril. Doi: 10.1007 / 164_2020_361.
5 Una revisión de las terapias basadas en genes y células para la hipercolesterolemia familiar. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A. Pharmacol Res. Mayo de 2019; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.