

Síndrome de QT largo: causas, diagnóstico, valores, tratamiento, medicación
El síndrome de QT largo (SQTL) hace referencia a un conjunto de síntomas causados por una anomalía cardiaca caracterizada por un retraso en la repolarización de las células miocárdicas y asociada a síncope (desmayo con pérdida de conciencia y tono postural)
El síncope es causado con mayor frecuencia por arritmias malignas, especialmente torsiones de la punta, que pueden degenerar en fibrilación ventricular, lo que lleva a un paro cardíaco irreversible de la persona afectada (muerte cardíaca súbita).
Las arritmias en pacientes con LQTS a menudo se desencadenan por el ejercicio o por estímulos emocionales muy fuertes, como el terror.
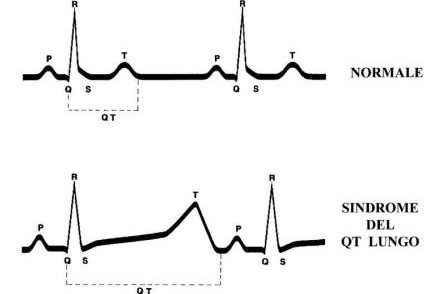
Las personas con LQTS tienen una prolongación característica del intervalo QT en el electrocardiograma: este intervalo se mide desde el comienzo de la onda Q hasta el final de la onda T (ver imagen arriba).
Detrás de todas las formas de LQTS hay una repolarización anormal del miocardio.
Las anomalías en la repolarización provocan diferencias en la refractariedad del miocardio.
Debido a estas diferencias, cualquier posdespolarización (que ocurre con mayor frecuencia en pacientes con LQTS) puede propagarse a las células adyacentes, lo que provoca arritmias ventriculares de reentrada.
Epidemiología y factores de riesgo en el síndrome de QT largo (SQTL)
En individuos genéticamente predispuestos, los aumentos repentinos del tono simpático, como los que pueden ocurrir durante un esfuerzo excesivo o emociones violentas, son factores de riesgo para la aparición de arritmias malignas.
El síndrome afecta predominantemente a mujeres jóvenes. Entre los afectados, la incidencia de torsión de punta, síncope y muerte súbita cardiaca es mayor en caso de sordera congénita, taquiarritmia previa o síncope; también aumenta en proporción al alargamiento del intervalo QT.
Se estima que el riesgo relativo de arritmias malignas aumenta entre 1.1 y 1.2 veces por cada 10 ms de prolongación del intervalo QTc por encima de los valores normales.
Variantes clínicas
Distinguimos dos síndromes congénitos diferentes caracterizados por prolongación del intervalo QT y riesgo de muerte súbita por arritmias ventriculares:
- Síndrome de Romano-Ward, que se hereda según el modelo autosómico dominante (no asociado a sordera neurológica congénita u otras cardiopatías congénitas, autismo, sindactilia completa e inmunodeficiencia).
- Síndrome de Jervell-Lange-Nielsen, que se hereda con patrón autosómico recesivo (asociado a sordera neurológica congénita u otras cardiopatías congénitas, autismo, sindactilia completa e inmunodeficiencia).
Causas del LQTS
Hay dos formas de LQTS: congénita (más rara) y adquirida (más frecuente).
formas adquiridas
La mayoría de los casos vistos en la práctica clínica son formas adquiridas, que se pueden dividir en dos categorías: las generadas por trastornos del equilibrio hidroelectrolítico y las dependientes de la administración de fármacos.
Formas inducidas por trastornos electrolíticos.
- hipopotasemia
- hipomagnesemia
- hipocalcemia
- Formas inducidas por fármacos
- drogas antiarrítmicas
- La quinidina
- La amiodarona
- Sotalol
- Procainamida
- Ranolazine
- Antihistamínicos
- terfenadina
- astemizol
- mizolastina
- Antibióticos macrólidos
- Eritromicina
- Ciertos antibióticos de fluoroquinolona
- Ansiolíticos mayores
- Antidepresivos tricíclicos
- Agentes activos sobre la motilidad gastrointestinal
- La cisaprida
- Domperidona
- Drogas antipsicóticas
- Haloperidol
- La quetiapina
- La tioridazina
- Droperidol
- Analgésicos
- Metadona
- me LAAM
Al igual que las formas congénitas de LQTS, las formas adquiridas pueden provocar arritmias potencialmente mortales.
El tratamiento consiste en corregir el desequilibrio electrolítico, resolver su causa y suspender la terapia con el fármaco indicativo de prolongación del intervalo QT.
Dado su amplio uso, la tendencia a las interacciones con otros fármacos y la capacidad inherente de provocar la prolongación del intervalo QT, la eritromicina es probablemente la causa predominante del síndrome de QT prolongado adquirido.
De hecho, el uso de eritromicina está asociado con una incidencia de muerte súbita de más del doble que la de otros antibióticos.
Además de las dos categorías principales enumeradas anteriormente, debe recordarse que existen otras causas de prolongación del intervalo QT, como la anorexia nerviosa, el hipotiroidismo, la infección por VIH, la miocarditis y el infarto de miocardio.
formas congénitas
Las formas congénitas de LQTS pueden determinarse por mutaciones en uno de varios genes identificados hasta la fecha.
Estas mutaciones tienden a prolongar la duración del potencial de acción ventricular (APD), alargando así el intervalo QT.
Las formas congénitas se pueden heredar como un carácter autosómico dominante o autosómico recesivo.
La forma autosómica recesiva se asocia con otras cardiopatías congénitas, autismo, sindactilia completa e inmunodeficiencia (LQTS8) o con sordera neurológica congénita (LQTS1).
Se está identificando un número cada vez mayor de loci de genes en asociación con LQTS. Las pruebas genéticas para LQTS están disponibles en la práctica clínica y también pueden ayudar a establecer la terapia adecuada (Descripción general de las pruebas genéticas de LQTS).
Las causas más comunes de LQTS involucran mutaciones en los genes KCNQ1 (LQTS1), KCNH2 (LQTS2) y SCN5A (LQT3).
Síndrome de Jervell y Lange-Nielsen
Es la forma autosómica recesiva de LQTS.
Se asocia con sordera neurológica congénita u otras cardiopatías congénitas, autismo, sindactilia completa e inmunodeficiencia.
Está causada específicamente por una mutación en los genes KCNE1 y KCNQ1.
De las personas no tratadas, aproximadamente el 50% muere antes de los 15 años debido a arritmias ventriculares.
Síndrome de Romano-Ward
El síndrome de Romano-Ward es la forma autosómica dominante de LQTS.
No se asocia con sordera neurológica congénita u otras cardiopatías congénitas, autismo, sindactilia completa e inmunodeficiencia.
Diagnóstico y valores del Síndrome de QT Largo:
El diagnóstico de LQTS a menudo no es fácil ya que el 2.5% de la población sana tiene un QT prolongado y el 10-15% de los pacientes con LQTS tienen un intervalo QT normal: a menudo, incluso algunos atletas profesionales pueden tenerlo sin que el personal médico lo note.
Un criterio de diagnóstico de uso común se basa en la "puntuación de diagnóstico" de LQTS.

La puntuación se calcula asignando puntos de acuerdo con varios criterios que se enumeran a continuación.
Con 4 o más puntos la probabilidad de LQTS es alta y con 1 punto o menos la probabilidad es baja; 2 o 3 puntos indican una probabilidad intermedia.
QTc (definido como intervalo QT/raíz cuadrada del intervalo RR)
- >= 480 ms – 3 puntos
- 460-470 mseg – 2 puntos
- 450 mseg y género masculino – 1 punto
- Taquicardias tipo torsades de pointes ventriculares – 2 puntos
- Alternancia de onda T – 1 punto
- Avalancha de onda T en al menos 3 derivaciones en ECG - 1 punto
- Frecuencia cardíaca baja para la edad (niños): 0.5 puntos
- Síncope (no se pueden otorgar puntos por síncope y torsión máxima en el mismo sujeto)
- Bajo condiciones de estrés – 2 puntos
- Fuera de condiciones de estrés – 1 punto
- Sordera congénita – 0.5 puntos
- Antecedentes familiares (el mismo miembro de la familia no se puede contar tanto para la muerte súbita como para el LQTS)
- Otros miembros de la familia con diagnóstico definitivo de LQTS - 1 punto
- Muerte súbita en familiares cercanos (miembros menores de 30 años) – 0.5 puntos
Terapia
En pacientes asintomáticos sin demostración de arritmias ventriculares y en ausencia de antecedentes familiares positivos de muerte súbita, se recomienda observación sola y posiblemente terapia farmacológica sin necesidad de ir a la dosis máxima tolerada.
En LQTS1 y LQTS2, se pueden usar bloqueadores beta; en LQTS 3, son preferibles los antiarrítmicos de clase Ib como la mexiletina.
Los mismos medicamentos se pueden usar para tratar pacientes de emergencia, con la advertencia de que solo se debe usar lidocaína hasta que se confirme el diagnóstico de LQTS1 o LQTS2, ya que los betabloqueantes pueden empeorar las arritmias en pacientes con LQTS3.
Sin embargo, la terapia antiarrítmica debe adoptarse y realizarse a la dosis máxima tolerada en aquellos pacientes asintomáticos pero con evidencia de arritmias ventriculares no sostenidas y antecedentes familiares de muerte súbita.
Cardioversor implantable desfibrilador (ICD) la implantación no se recomienda estrictamente en estos pacientes.
Sin embargo, este último está absolutamente indicado (indicación de clase I) en pacientes con síncope o que ya han tenido un episodio de paro cardíaco.
La función de marcapasos del dispositivo debe ser aprovechada en aquellos que presentan arritmias durante la bradicardia o pausas en el ritmo cardíaco.
En pacientes que aún presentan síntomas a pesar de la terapia médica óptima, está indicada la cirugía de gangliectomía cérvico-torácica izquierda, con destrucción del ganglio estrellado y los tres o cuatro primeros ganglios simpáticos torácicos.
Pronóstico y riesgo
Para pacientes con LQTS no tratados, el riesgo de un evento (síncope o paro cardíaco) se puede estimar conociendo su genotipo (LQTS1-10), sexo e intervalo QTc.
Alto riesgo (>50%)
QTc>500 mseg LQTS1 & LQTS2 & LQTS3 (hombres)
Riesgo intermedio (30-50%)
QTc>500 mseg LQTS3 (mujeres)
QTc<500 mseg LQTS2 (mujeres) y LQTS3
Bajo riesgo (<30%)
QTc<500 mseg LQT1 y LQT2 (hombres)
Síndrome de QT largo y deporte
Los pacientes con LQTS, bajo estricto control cardiológico, aún pueden practicar deportes, pero eviten aquellos que incluyan un gran esfuerzo prolongado y deportes acuáticos como la natación y el buceo.
Lea también:
Enfermedad cardíaca: ¿Qué es la miocardiopatía?
Inflamaciones del corazón: miocarditis, endocarditis infecciosa y pericarditis
Soplos cardíacos: qué es y cuándo preocuparse
El síndrome del corazón roto está en aumento: conocemos la miocardiopatía de Takotsubo
¿Qué es un cardioversor? Descripción general del desfibrilador implantable
Inflamaciones del corazón: ¿Cuáles son las causas de la pericarditis?
¿Tiene episodios de taquicardia repentina? Puede sufrir del síndrome de Wolff-Parkinson-White (WPW)
Saber que la trombosis interviene en el coágulo de sangre
Procedimientos del paciente: ¿Qué es la cardioversión eléctrica externa?
Aumentar la fuerza laboral de EMS, capacitar a laicos en el uso de AED
Diferencia entre cardioversión espontánea, eléctrica y farmacológica
¿Qué es la miocardiopatía de Takotsubo (síndrome del corazón roto)?
El ECG del Paciente: Cómo Leer un Electrocardiograma de Manera Sencilla
Prueba de esfuerzo que induce arritmias ventriculares en individuos con intervalo de LQT