
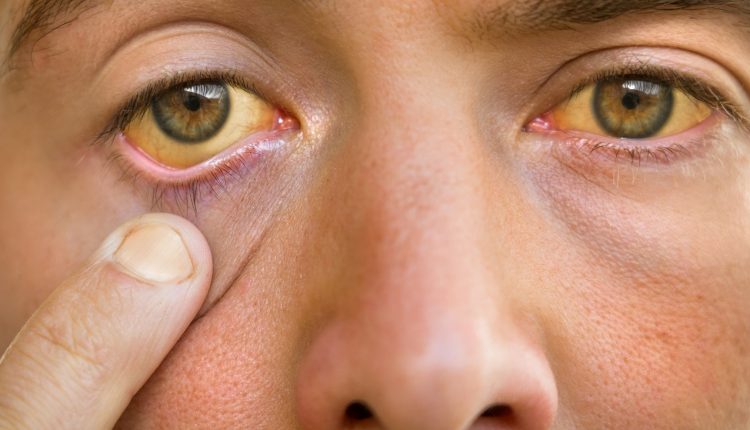
Che cos'è la Sindrome di Gilbert
La sindrome di Gilbert è una condizione fisiopatologica familiare ed ereditaria, di scarso rilievo clinico e con prognosi benigna, caratterizzata da ittero o subittero epatico, causata da una congenita alterazione del metabolismo della bilirubina e conseguente aumento della bilirubinemia nel sangue, ovvero presenza di iperbilirubinemia (con ittero o subittero sclerale)
Le iperbilirubinemie si manifestano, infatti, con ittero e si differenziano secondo varie e diverse tipologie.
Gli itteri si classificano in tre specie: pre-epatico, epatico, post epatico.
La sindrome di Gilbert è un ittero (o subittero se meno manifesto) di tipo epatico, con variabile intensità
Dipende da una alterata funzione all’interno dell’epatocita, la cellula principale del fegato; si tratta di un difetto enzimatico, congenito e familiare, che comporta una disfunzione e riduzione del normale processo di glucurono-coniugazione della bilirubina, con conseguente eccesso di bilirubina indiretta nel siero e nella composizione della bile.
La bilirubina è il prodotto finale della normale degradazione dell’eme, proviene dal catabolismo dell’eme contenuto nel globulo rosso senescente (ogni globulo rosso ha una durata di circa 100 giorni, nel Gilbert anche meno); la produzione della bilirubina avviene nel sistema reticolo-endoteliale, soprattutto milza e midollo osseo, dopodiché viene captata dagli epatociti nel fegato che la sottopongono al processo di glucurono-coniugazione che nella sindrome di Gilbert è difettoso.
Per cui la bilirunina viene rilasciata in forma indiretta o “non coniugata”.
Tale disordine del metabolismo della bilirubina può avere diversi gradi di gravità, da grado modesto come nella sindrome di Gilbert, a grado severo come nella iperbilirubinemia elevata delle sindromi di Crigler-Najjar I e II, fortunatamente meno frequenti e rare.
Come si manifesta la Sindrome di Gilbert
La sindrome di Gilbert è una condizione abbastanza frequente (circa 5 – 8% della popolazione italiana).
I soggetti che ne sono affetti presentano nel siero un aumento, di solito fluttuante, della bilirubinemia, soprattutto della bilirubinemia indiretta o non coniugata (talvolta anche di quella diretta ma, nel caso, non prevalente).
Il tasso di bilirubinemia, e della bilirubina non coniugata, è generalmente molto variabile, e spesso è nella norma in tanti soggetti con sindrome di Gilbert.
Si tratta di un’alterazione a carattere ereditario che non crea alcun tipo di problema sia sulla qualità, sia sulla durata di vita dei soggetti che ne sono affetti.
In alcune condizioni (febbre, infezioni, intercorrenti, stress, abuso di alcool, sforzi fisici, digiuno) si può avere un netto incremento, anche sopra il valore di 2,5 – 5 mg/100 ml della bilirubinemia che tuttavia ritorna prontamente nella norma quando cessano queste situazioni particolari, di solito momentanee e provvisorie.
Diagnosi della Sindrome di Gilbert
La sindrome di Gilbert viene scoperta nel soggetto per lo più casualmente, per lo più nel corso di indagini di laboratorio eseguite per controlli di routine o occasionalmente.
L’età di insorgenza di un primo episodio itterico o subitterico è di solito intorno ai 15-18 anni.
Nel 40% dei soggetti con sindrome di Gilbert è presente una lieve diminuzione della vita media dei globuli rossi, che può indurre un sospetto di emolisi.
Per la diagnosi di sindrome di Gilbert si richiede preliminarmente l’esclusione di ogni altra malattia epatocellulare, biliare, emolitica che possa causare (o concausare) ittero; inoltre occorre il riscontro di un aumento della bilirubinemia, prevalentemente indiretta per almeno tre volte successive, due o tre rilievi di incremento di valori sierici riscontrati ad intervalli di alcuni mesi, in concomitanza di assoluta normalità di tutti i test di funzionalità epatica (come transaminasi, gGT, fosfatasi alcalina, bilirubinemia diretta e indiretta, acidi biliari, emocromo) e cioè la presenza di una iperbilirubinemia isolata.
Può essere anche utile e necessaria una ecografia addominale nella norma che accerti la normalità del fegato e delle vie biliari intra ed extraepatiche.
È molto difficile che si effettui una biopsia epatica per una tale diagnosi, se non forse nel caso di dover ricercare una più approfondita diagnosi differenziale in situazioni problematiche o intricate.
La diagnosi differenziale, per distinguere la sindrome di Gilbert da altre patologie che presentano qualche caratteristica comune, va fatta soprattutto con le altre patologie ereditarie che riguardano il metabolismo della bilirubina e cioè, come accennato prima, la sindrome di Crigler-Najjar I, molto più grave e con prognosi infausta; la sindrome di Crigler-Najar II, meno preoccupante e con prognosi relativamente buona ma con valori di iperbilirubinemia indiretta più alti del Gilbert e insorgenza più precoce, per lo più in età infantile, a differenza del Gilbert, in cui l’esordio dell’ittero compare di solito più tardivamente, cioè intorno ai 15-18 anni.
La sindrome di Rotor e la sindrome di Dubin-Johnson sono altrettanto iperbilirubinemie familiari ed ereditarie ma con aumento della bilirubina diretta o coniugata
Sono anch’esse patologie del metabolismo della bilirubina, condizioni molto rare, ma con tutt’ altra disfunzione, con meccanismi patogenetici diversi, anche con ittero più intenso; occasionalmente si possono trovare anche in gravidanza o durante una terapia con estroprogestinici, talvolta da distinguere dall’ittero di tipo post-epatico, ma con indici di colestasi normali.
È opportuno ricordare altre forme di condizioni morbose caratterizzate da aumento della bilirubinemia non coniugata (o indiretta) nel sangue come nella sindrome di Gilbert: l’ittero fisiologico del neonato e l’ittero da latte materno, che si manifestano alla nascita, di natura benigna e momentanea. Si presenta, rispettivamente, un ritardo di maturazione del metabolismo della bilirubina e, nel secondo caso, la presenza di prolungamento dell’ittero fisiologico per una provvisoria alterazione della glicuronoconiugazione per predisposizione e con alterazioni genetiche simili a quelle della sindrome di Gilbert.

Altre condizioni di alterato metabolismo della bilirubina sono, inoltre, le iperbilirubinemia da farmaci (es. sulfonamide – un batteriostatico – o l’ acido fusidico – un antibiotico.)
Recentemente sono state descritte diverse forme di danno epatico in pazienti con iperbilirubinemia familiare, in particolare nel Gilbert; questi pazienti andrebbero strettamente monitorati dal punto di vista della funzione epatica quando necessitano di terapie con numerosi farmaci, o in caso di chemioterapia.
Infine, per completezza, relativamente soprattutto all’ittero, è ovviamente da ricordare tutto il grande capitolo delle Epatopatie acute e croniche (le epatiti), le ostruzioni dell’albero biliare, e tutto il capitolo della Colestasi, per ciò che riguarda la bile, la secrezione biliare, le condizioni di coinvolgimento dell’epatocita, e da ultimo il grande capitolo dell’emolisi, in cui si trova iperbilirubinemia insieme alle alterazioni dei vari parametri ematochimici ematologici.
Prognosi della Sindrome di Gilbert
La prognosi della sindrome di Gilbert è, come si è detto, buona ed anche eccellente.
Non vi sono, infatti, effetti nocivi sulla funzione epatica né sullo stato di benessere dell’intero organismo.
Terapia per la Sindrome di Gilbert
Non è necessaria alcuna terapia.
Come misura per evitare eventuali incrementi temporanei della iperbilirubinemia, si consiglia di:
- ridurre o prevenire stress
- evitare il digiuno
- non abusare con gli alcoolici
- evitare gli sforzi fisici eccessivi.
Per approfondire:
Emergency Live ancora più…live: scarica la nuova app gratuita del tuo giornale per iOS e Android
Sindrome di Gilbert: sintomi, cause e diagnosi di questa patologia del fegato
Epatite acuta nei bambini, Maggiore (Bambino Gesù): “Ittero campanello d’allarme”
Quali sono le cause dell’epatite A e come curarsi
Le diverse tipologie di epatite: prevenzione e cura
Casi di epatite acuta nei bambini: conosciamo le epatiti virali
Ipercolesterolemia familiare: cos’è e come trattarla
Steatosi Epatica: cause e cure del fegato grasso
Epatopatia: gli esami non invasivi per valutare la malattia epatica
Fegato: che cos’è la steatoepatite non alcolica