
Familiehyperkolesterolemi: hva det er og hvordan man behandler det
Familial hyperkolesterolemi, en sjelden genetisk sykdom som forårsaker alvorlige helsekonsekvenser (f.eks. Hjerteinfarkt og hjerneslag) som krever passende inneslutningstiltak med passende medisiner og en passende livsstil.
Hva betyr hyperkolesterolemi? Og hva betyr familiær?
Hyperkolesterolemi er en klinisk tilstand preget av overdreven kolesterol i blodet.
Kolesterol er en komponent av lipider, vanligvis tatt med dietten, men som også kan produseres av kroppen.
Kolesterol er veldig viktig for livet fordi det brukes til dannelse av cellemembraner (for å sikre deres funksjon), beskytter nevroner og hjernenerver.
Kolesterol brukes også til syntese av andre molekyler, hvorav tre er gallsyrer (viktig for fordøyelsen), noen hormoner og vitamin D.
Bare hvis det er over disse kravene, kan det forårsake alvorlig skade.
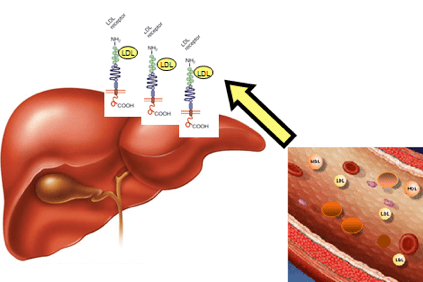
Når det gjelder hyperkolesterolemi, akkumuleres kolesterol i blodet i form av lette lipoproteiner (LDL), spesielle aggregater av fett og proteiner, også kalt ”dårlig kolesterol” som fremmer dannelsen av plakk i arterieveggen (aterosklerotiske plakk).
Dette skjer når pasienter har så høye kolesterolnivåer at de ikke kan eliminere det gjennom leverens fysiologiske mekanismer.
Akkumuleringen av LDL i blodet fører til slutt til dannelsen av atheroma (en fysisk hindring for normal blodstrøm) som kan føre til mer alvorlige konsekvenser som angina, hjerteinfarkt, hjerneslag, men også i andre organer som hjernen, nyrer, lunger og selve leveren.
Hyperkolesterolemi er kjent når den overføres til avkom
Dette skyldes modifikasjoner av et gen som inneholder informasjonen for å lage et leverprotein, LDLR (LDLR) -reseptoren, som gjenkjenner LDL, fjerner det fra blodet og transporterer det til levercellene, som deretter fjerner det. Denne genendringen forårsaker LDL-akkumulering i blodet.
Til dags dato er det mer enn 600 kjente endringer i LDL-genet som forårsaker hyperkolesterolemi i familien.
Også personer med denne lidelsen kan føre til endringer i andre gener som er involvert i lipoproteinmetabolismer som APOB-genet (apoprotein B i LDL) og PCSK9-genet (et protein som nedbryter LDL-reseptorer).
Selv om det er kjent at kolesterolakkumulering kan være skadelig, anslås det at opptil en tredjedel av hjerteinfarkt som oppstår før fylte 40 år skyldes utiagnostisert eller upassende behandlet familiehyperkolesterolemi.
Mer vanlig er multifaktoriell hyperkolesterolemi forårsaket av miljøfaktorer (et kosthold med høyt fettinnhold, spesielt hvis det er forbundet med fysisk inaktivitet) selv i nærvær av predisponerende genetiske faktorer.
Formene som familie hyperkolesterolemi manifesterer seg
Denne sykdommen kan presentere seg i to forskjellige former: en mindre alvorlig (heterozygot, 2 tilfelle hver 1 individer) og en mer alvorlig (homozygot, 500 tilfelle hver 1 individer) 1,000,000.
Den heterozygote formen er ofte asymptomatisk og diagnostiseres bare basert på kolesterolnivået i blodet.
Leveren sliter med å eliminere LDL fordi LDL-reseptorer produseres i utilstrekkelig antall, noe som resulterer i en 2 eller 3 ganger økning i blodnivået sammenlignet med normale verdier.
Denne formen kan føre til økt risiko for hjerte- og karsykdommer i voksen alder.
Den homozygote formen er preget av utbruddet av kardiovaskulær sykdom selv i ung alder og tilstedeværelsen av karakteristiske opphopninger av fett som xanthomas (gulaktige knuter på hendene og akillessenen) og xanthelasma (gullige plater på øyelokkene og rundt øynene).
Den genetiske defekten arves fra begge foreldrene, og risikoen for hjerteinfarkt i fravær av terapi er allerede observert rundt 15-20 år.
Faktisk, i denne tilstanden, mislykkes leveren i å metabolisere lipoproteinene som er igjen i blodet, som deretter akkumuleres og føre til dysfunksjonene beskrevet ovenfor og skape en situasjon som er uforenlig med livet.
Hva er sjansene for overføring av sykdommen til barn?
Siden hver av oss har to kopier av hvert gen, hver arvet fra en forelder, vil vi ha den heterozygote formen når vi arver en endret og en sunn kopi.
Tvert imot, vi vil ha den homozygote formen hvis vi arver begge kopiene av det syke genet fra begge foreldrene. Det må sies at denne formen er svært sjelden fordi den bare oppstår når begge foreldrene har genet som forårsaker sykdommen, og derfor kan hver av dem overføre en kopi av den samme genendringen.
Familiær hyperkolesterolemi kalles heterozygot patologi sammensatt i de sjeldne tilfellene hvor to forskjellige typer genendringer arves, en fra hver av foreldrene.
Barn med risiko for å utvikle sykdommen må diagnostiseres og behandles tidlig. I dag er det mulig å diagnostisere sykdommen gjennom genetiske tester som ser etter feil (genendringer) på LDLR-, ApoB- og PCSK9-gener.
Derfor er bare formene for familiehyperkolesterolemi overførbare.
Imidlertid kan multifaktorielle former som hovedsakelig oppstår på grunn av en feil livsstil (akkumulering av kolesterol med dietten), også være preget av en genetisk disposisjon.
Faktisk kan disse fagene være til stede genetiske underskudd som kompromitterer kroppens evne til tilstrekkelig å kompensere for overflødig lipid i dietten.
For eksempel når leveren er mettet med kolesterol tatt med mat, kan produksjonen av reseptorer som fanger opp LDL som sirkulerer i blodet, undertrykkes.
En situasjon er veldig lik den som oppstår under hyperkolesterolemi i familien, selv om den er mindre alvorlig.
I dette tilfellet er konsentrasjonen av totalt kolesterol i blodet over det normale og er vanligvis mellom 240 og 350 mg / dl sammenlignet med det normale som vanligvis er satt til 200 - 240 mg / dl.

En nysgjerrighet: denne sykdommen kan ha blitt overlevert gjennom århundrene
En nøye observasjon av det berømte maleriet av Mona Lisa viser muligheten for at Mona Lisa allerede i ung alder led av familiehyperkolesterolemi.
Faktisk skildrer Leonardo også trofast de typiske fettavsetningene på hendene og nær øynene (nå kjent som xanthomas og xanthelasma) som indikerer tilstedeværelsen av patologien med sikkerhet.
Hvis dette stemmer, er det vist at hyperkolesterolemi i familien er en sykdom bevart gjennom århundrene (en sak dokumentert allerede i 1500).
Hva er mulighetene for å behandle denne sykdommen i dag?
Muligheten for å kurere sykdommen avhenger av alvorlighetsgraden. Risikofaktorer (diett, røyking, alder, familie og personlig historie med hyperkolesterolemi, tilstedeværelse av andre sykdommer) kan også forverre det samlede sykdomsbildet.
For å behandle hyperkolesterolemi i familien kan nye generasjons medisiner brukes, men samtidig er det nødvendig å handle med en passende livsstilsendring.
I tilfeller av middels alvorlighetsgrad (heterozygot familiehyperkolesterolemi), statinbasert medisinering (kolesterolsyntesehemmere som induserer økt LDLR-reseptorsyntese) og kombinasjonen med ezetimib (kolesterolabsorpsjonshemmere) eller PCSK9-hemmende medisiner (dvs. PCSK9-proteinhemmere hvis rolle er å ødelegge leverreseptorer som fanger opp LDLR) forbedrer aktiviteten til den sunne kopien av LDLR-genet og reduserer akkumuleringen av kolesterol i blodet.
Nylig godkjent er også bempedoesyre, et medikament som virker i leveren ved å hemme enzymet ATP citratlyase, et molekyl som er involvert i prosessen med endogen kolesterolsyntese.
Denne virkningsmekanismen tillater oss å handle på mengden produsert kolesterol, som virker oppstrøms fra statinens virkningssted, og å stimulere ekspresjon av LDL-reseptorer for å kompensere for den reduserte syntesen.
I motsetning til statiner er bempedoesyre ikke aktiv i skjelettmuskulaturen, noe som reduserer muligheten for å ha uønskede hendelser som er typiske for statiner.
Dette legemidlet kan også assosieres med ezetimib med gunstige effekter.
Homozygot familiær hyperkolesterolemi ble inntil nylig ansett som en uhelbredelig sykdom.
Statinbasert terapi er ikke effektiv i denne patologien. Faktisk kan statiner som virker på mekanismene som fører til produksjon av endogent kolesterol ikke stimulere syntesen av LDL-reseptorer.
For å eliminere LDL-kolesterol fra kroppen til disse pasientene, er plasmaferese, en teknikk som gjør det mulig å filtrere blodet ved å eliminere fett, på samme måte som det som gjøres med dialyse når nyrene ikke fungerer.
Dette er imidlertid en invasiv prosedyre med negativ innvirkning på pasientenes livskvalitet.
Den nyeste forskningen på familiehyperkolesterolemi
I de senere år har forskning ført til utvikling av spesifikke medikamenter også for den homozygote formen av familiehyperkolesterolemi, noe som forbedrer forventningene og livskvaliteten til pasienter som lider av det.
For eksempel fører medikamentet lomitapid (tatt oralt) til markant lavere plasma LDL-kolesterolnivå hos disse pasientene.
Lomitapid hemmer et mikrosomalt triglyseridtransportprotein (MTP) som tillater inkorporering av disse lipidene sammen med apoproteinet B100 i fremvoksende VLDL.
Som et resultat reduseres apoproteinet B100 og LDL selv hos pasienter der sykdommen har forårsaket det totale fraværet av LDL-reseptoren.
En annen ny generasjons medikament er mipomersen, et antisense-oligonukleotid som er i stand til å nedbryte B100-apoproteinene som deltar i dannelsen av LDL, og dermed redusere antallet sistnevnte.
Evolocumab og alirocumab er to monoklonale antistoffer som hemmer aktiviteten til PCSK9-proteinet i blodet.
Men for at disse to legemidlene skal tre i kraft, må minst en liten del av LDL-reseptorene være tilstede og fungere.
Blant legemidlene som fremdeles er i klinisk utvikling er inkluderingen av et biologisk legemiddel (såkalt siRNA) som blokkerer DNA-transkripsjon og syntese av PCSK9-proteinet i leveren.
Det er nylig oppnådd positive resultater med evinacumab, en monoklonal antistoffhemmere av proteinet lignende-angiopoietin 3 (ANGPTL3), et molekyl syntetisert av leveren hvis rolle er å øke kolesterol-LLDL og triglyseridnivåer som forhindrer nedbrytning.
Dette nye legemidlet har også vist effekt hos pasienter der LDL-reseptoren ikke fungerer3,4.
Nylig har det også blitt gjort noen forsøk på å forstå om genterapi, med introduksjonen av det sunne genet i pasientens DNA, kunne fungere5.
Fra denne listen over mulige terapier for familiær hyperkolesterolemi er det mulig å forstå hvor mye forskning som gjøres for å gi løsninger for disse pasientene som, selv om de er sjeldne, er rammet av denne alvorlige sykdommen, noe som reduserer risikoen for dødelige konsekvenser.
Imidlertid, som alltid skjer med nye legemidler, er det bra å bruke et ord med forsiktighet ved kronisk bruk, siden kunnskapen om deres mulige bivirkninger bare vil være tydeligere med erfaring (legemiddelovervåkning), så bruken av dem i barn bør vurderes nøye, selvfølgelig med det endelige målet å tilby disse "små" pasientene muligheten til å leve et "normalt" liv.
Bibliografiske og sitografiske referanser for artikkelen om hyperkolesterolemi i familien
1 2019 ESC / EAS-retningslinjer for behandling av dyslipidemi: Lipidmodifisering for å redusere kardiovaskulær risiko. Forfattere / Task Force Members; ESC Committee for Practice Guidelines (CPG); ESC National Cardiac Societies. Åreforkalkning. 2019 nov; 290: 140-205. doi: 10.1016 / j.aterosklerose.2019.08.014.
2 Sjeldne dyslipidaemier, fra fenotype til genotype til ledelse: en konsensusuttalelse fra European Atherosclerosis Society. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet Diabetes Endocrinol. 2020 jan; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoprotein (a) Senking fra lipoproteinaferese til antisense-oligonukleotid-tilnærming. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 3. juli; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 LDL-kolesterolsenkende terapi. Pirillo A, Norata GD, Catapano AL. Handb Exp Pharmacol. 2020 30. april. Doi: 10.1007 / 164_2020_361.
5 En gjennomgang av gen- og cellebaserte terapier for familiær hyperkolesterolemi. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A.Pharmacol Res. 2019 mai; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.