
Perheen hyperkolesterolemia: mikä se on ja miten sitä hoidetaan
Perheellinen hyperkolesterolemia, harvinainen geneettinen sairaus, joka aiheuttaa vakavia terveysvaikutuksia (esim. Sydänkohtaus ja aivohalvaus), joka vaatii asianmukaisia suojatoimenpiteitä asianmukaisella lääkityksellä ja sopivalla elämäntavalla.
Mitä hyperkolesterolemia tarkoittaa? Ja mitä perhe tarkoittaa?
Hyperkolesterolemia on kliininen tila, jolle on ominaista veren kolesteroli.
Kolesteroli on lipidien komponentti, joka otetaan yleensä ruokavalion kanssa, mutta jota elimistö voi myös tuottaa.
Kolesteroli on erittäin tärkeä elämälle, koska sitä käytetään solukalvojen muodostumiseen (niiden toiminnan varmistamiseksi), suojaa hermosoluja ja kallonhermoja.
Kolesterolia käytetään myös muiden molekyylien synteesiin, joista kolme on sappihappoja (tärkeä ruoansulatukselle), joitain hormoneja ja D-vitamiinia.
Ainoastaan näiden vaatimusten yläpuolella oleva se voi aiheuttaa vakavia vahinkoja.
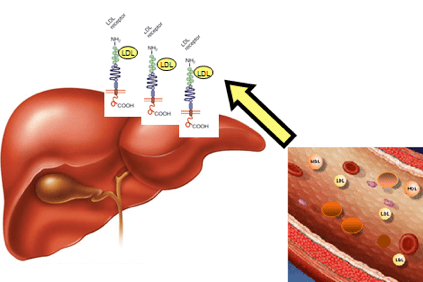
Hyperkolesterolemian tapauksessa kolesteroli kerääntyy vereen kevyiden lipoproteiinien (LDL) muodossa, erityisesti rasvojen ja proteiinien aggregaattien, joita kutsutaan myös "huonoksi kolesteroliksi", jotka edistävät plakkien muodostumista valtimon seinämässä (ateroskleroottiset plakit).
Tämä tapahtuu, kun potilailla on niin korkea kolesterolitaso, että he eivät voi poistaa sitä maksan fysiologisten mekanismien kautta.
LDL: n kertyminen vereen johtaa lopulta aterooman muodostumiseen (fyysinen este normaalille verenvirtaukselle), mikä voi johtaa vakavampiin seurauksiin, kuten angina pectoris, sydänkohtaus, aivohalvaus, mutta myös muihin elimiin, kuten aivoihin, munuaiset, keuhkot ja itse maksa.
Hyperkolesterolemia on tuttua, kun se siirretään jälkeläisille
Tämä johtuu muutoksista geenissä, joka sisältää tiedot maksaproteiinin valmistamiseksi, LDLR (LDLR) -reseptori, joka tunnistaa LDL: n, poistaa sen verenkierrosta ja kuljettaa maksasoluihin, jotka sitten poistavat sen. Tämä geenimuutos aiheuttaa LDL: n kertymisen veressä.
Tähän mennessä LDL-geenissä on yli 600 tunnettua muutosta, jotka aiheuttavat perheen hyperkolesterolemiaa.
Myös ihmiset, joilla on tämä häiriö, voivat tuoda muutoksia muihin lipoproteiinimetabolioihin liittyviin geeneihin, kuten APOB-geeni (apoproteiini B LDL: ssä) ja PCSK9-geeni (proteiini, joka hajottaa LDL-reseptoreita).
Vaikka tiedetään, että kolesterolin kerääntyminen voi olla haitallista, arvioidaan, että jopa kolmasosa ennen 40-vuotiaita esiintyvistä sydänkohtauksista johtuu diagnosoimattomasta tai epäasianmukaisesti hoidetusta perheen hyperkolesterolemiasta.
Yleisempää on ympäristötekijöiden aiheuttama monitekijäinen hyperkolesterolemia (runsaasti rasvaa sisältävä ruokavalio, varsinkin jos se liittyy fyysiseen passiivisuuteen) jopa altistavien geneettisten tekijöiden läsnä ollessa.
Muotot, jotka perheen hyperkolesterolemia ilmenee
Tämä tauti voi esiintyä kahdessa eri muodossa: vähemmän vakava (heterotsygoottinen, yksi tapaus jokaista 2 yksilöä kohti) ja vakavampi (homotsygootti, yksi tapaus 1 500 1 yksilöä kohti) 1,000,000.
Heterotsygoottinen muoto on usein oireeton ja se diagnosoidaan vain veren kolesterolitasojen perusteella.
Maksa kamppailee LDL: n poistamiseksi, koska LDL-reseptoreita tuotetaan riittämättömässä määrin, mikä johtaa veritasojen nousuun 2 tai 3 kertaa normaaliarvoihin verrattuna.
Tämä muoto voi johtaa lisääntyneeseen sydän- ja verisuonitautien riskiin aikuisiässä.
Homotsygoottiselle muodolle on tunnusomaista sydän- ja verisuonitautien puhkeaminen jo nuorena ja tyypillisten rasvakertymien, kuten ksantoomien (kellertävät kyhmyt käsien ja Achilles-jänteen solmissa) ja ksantelasman (kellertävät levyt silmäluomissa ja ympärillä) läsnäolo. silmät).
Geneettinen vika periytyy molemmilta vanhemmilta, ja sydänkohtauksen riski hoidon puuttuessa havaitaan jo noin 15-20 vuoden iässä.
Itse asiassa tässä tilassa maksa ei pysty metabolisoimaan veressä olevia lipoproteiineja, jotka sitten kertyvät johtamaan yllä kuvattuihin toimintahäiriöihin ja luovat elämän kanssa yhteensopimattoman tilanteen.
Mitkä ovat taudin tarttumismahdollisuudet lapsille?
Koska kullakin meistä on kaksi kopiota kustakin geenistä, kukin peritty vanhemmilta, meillä on heterotsygoottinen muoto, kun perimme yhden muutetun ja yhden terveellisen kopion.
Päinvastoin, meillä on homotsygoottinen muoto, jos perimme sairaan geenin molemmat kopiot molemmilta vanhemmilta. On sanottava, että tämä muoto on hyvin harvinainen, koska se esiintyy vain, kun molemmilla vanhemmilla on taudin aiheuttava geeni, ja siksi kukin heistä voi lähettää kopion samasta geenimuutoksesta.
Perheellistä hyperkolesterolemiaa kutsutaan heterotsygoottiseksi patologiaksi, joka koostuu harvoista tapauksista, joissa peritään kaksi erityyppistä geenimuutosta, yksi kustakin vanhemmasta.
Lapset, joilla on taudin kehittymisen riski, on diagnosoitava ja hoidettava aikaisin. Nykyään tauti on mahdollista diagnosoida geneettisillä testeillä, jotka etsivät virheitä (geenimuutoksia) LDLR-, ApoB- ja PCSK9-geeneissä.
Siksi vain perheen hyperkolesterolemian muodot ovat tarttuvia.
Kuitenkin monitekijöisille muodoille, jotka johtuvat pääasiassa väärästä elämäntavasta (kolesterolin kertyminen ruokavalion kanssa), voidaan myös luonnehtia geneettinen taipumus.
Itse asiassa näillä kohteilla voi olla geneettisiä puutteita, jotka vaarantavat kehon kyvyn kompensoida riittävästi ylimääräistä lipidiä ruokavaliossa.
Esimerkiksi kun maksa on kyllästetty ruoan kanssa otetulla kolesterolilla, veressä kiertävän LDL: n sieppaavien reseptorien tuotanto voidaan estää.
Tilanne on hyvin samanlainen kuin perheiden hyperkolesterolemian aikana, vaikkakin lievempi.
Tässä tapauksessa kokonaiskolesterolin pitoisuus veressä on normaalia korkeampi ja on yleensä välillä 240-350 mg / dl verrattuna normaaliin, joka on tavallisesti asetettu noin 200-240 mg / dl.

Uteliaisuus: tämä tauti on saatettu antaa vuosisatojen ajan
Mona Lisan kuuluisan maalauksen tarkka tarkkailu osoittaa, että Mona Lisa kärsi jo nuorena perheen hyperkolesterolemiasta.
Itse asiassa Leonardo kuvaa uskollisesti myös tyypillisiä rasvakertymiä käsissä ja silmien lähellä (nyt tunnetaan nimellä ksantoomat ja ksantelasmat), jotka osoittavat patologian läsnäolon varmuudella.
Jos tämä on totta, osoitetaan, että perheen hyperkolesterolemia on vuosisatojen ajan säilynyt sairaus (tapaus dokumentoitu jo vuonna 1500).
Mitkä ovat mahdollisuudet tämän taudin hoitamiseen nykyään?
Mahdollisuus parantaa tauti riippuu sen vakavuudesta. Myös riskitekijät (ruokavalio, tupakointi, ikä, perhe ja hyperkolesterolemian henkilökohtainen historia, muiden sairauksien esiintyminen) voivat pahentaa taudin kokonaiskuvaa.
Perheen hyperkolesterolemian hoidossa voidaan käyttää uuden sukupolven lääkkeitä, mutta samalla on tarpeen toimia sopivalla elämäntapamuutoksella.
Keskivaikeissa tapauksissa (heterotsygoottinen perheen hyperkolesterolemia) statiinipohjainen lääkehoito (kolesterolisynteesin estäjät, jotka indusoivat lisääntyneen LDLR-reseptorisynteesin) ja yhdistelmä ezetimibin (kolesterolin imeytymisen estäjät) tai PCSK9: n estäjien (eli PCSK9-proteiinin estäjien, joiden tehtävänä on tuhoavat LDLR: n sieppaavat maksareseptorit) parantaa LDLR-geenin terveellisen kopion aktiivisuutta ja vähentää kolesterolin kertymistä veressä.
Äskettäin hyväksytty on myös bempedoehappo, lääke, joka toimii maksassa estämällä entsyymiä ATP-sitraattilyaasia, molekyyliä, joka osallistuu endogeenisen kolesterolisynteesin prosessiin.
Tämän toimintamekanismin avulla voimme vaikuttaa tuotetun kolesterolin määrään, joka toimii ylävirtaan statiinien vaikutuspaikasta, ja stimuloida LDL-reseptorien ilmentymistä kompensoimaan alentunutta synteesiä.
Toisin kuin statiinit, bempedoiinihappo ei ole aktiivinen luurankolihaksissa, mikä vähentää statiinien tyypillisten ei-toivottujen tapahtumien mahdollisuutta.
Tähän lääkkeeseen voidaan liittää myös etsetimibiä, jolla on myönteisiä vaikutuksia.
Homotsygoottista familiaalista hyperkolesterolemiaa pidettiin viime aikoihin asti parantumattomana sairautena.
Statiinipohjainen hoito ei ole tehokasta tässä patologiassa. Itse asiassa statiinit, jotka vaikuttavat mekanismeihin, jotka johtavat endogeenisen kolesterolin tuotantoon, eivät voi stimuloida LDL-reseptorien synteesiä.
LDL-kolesterolin poistamiseksi näiden potilaiden kehosta plasmafereesi, tekniikka, joka mahdollistaa veren suodattamisen poistamalla rasvat, samoin kuin dialyysillä, kun munuaiset eivät toimi.
Tämä on kuitenkin invasiivinen menettely, jolla on negatiivinen vaikutus potilaiden elämänlaatuun.
Viimeisin tutkimus perheen hyperkolesterolemiasta
Viime vuosina tutkimus on johtanut spesifisten lääkkeiden kehittämiseen myös perheen hyperkolesterolemian homotsygoottiselle muodolle, mikä on parantanut sitä kärsivien potilaiden odotuksia ja elämänlaatua.
Esimerkiksi lomitapidi (oraalisesti) johtaa huomattavasti alempaan plasman LDL-kolesterolitasoon näillä potilailla.
Lomitapidi estää mikrosomaalisen triglyseridien kuljetusproteiinin (MTP), joka sallii näiden lipidien liittämisen yhdessä apoproteiini B100: n kanssa syntymässä oleviin VLDL: iin.
Tämän seurauksena apoproteiini B100 ja LDL vähenevät myös potilailla, joissa tauti on aiheuttanut LDL-reseptorin täydellisen puuttumisen.
Toinen uuden sukupolven lääke on mipomerseeni, antisense-oligonukleotidi, joka pystyy hajottamaan B100-apoproteiineja, jotka osallistuvat LDL: n muodostumiseen, vähentäen siten jälkimmäisten määrää.
Evolokumabi ja alirokumabi ovat kaksi monoklonaalista vasta-ainetta, jotka estävät PCSK9-proteiinin aktiivisuuden veressä.
Kuitenkin, jotta nämä kaksi lääkettä voisivat vaikuttaa, ainakin pienen osan LDL-reseptoreista on oltava läsnä ja toiminnassa.
Kliinisessä kehitysvaiheessa olevien lääkkeiden joukossa on biologisen lääkkeen (niin kutsutun siRNA: n) sisällyttäminen, joka estää DNA: n transkription ja PCSK9-proteiinin synteesin maksassa.
Positiivisia tuloksia on äskettäin saavutettu evinakumabilla, joka on proteiinin samanlainen-angiopoietiini 3 (ANGPTL3) monoklonaalinen vasta-aineinhibiittori, maksan syntetisoima molekyyli, jonka tehtävänä on lisätä kolesteroli-LLDL- ja triglyseriditasoja estäen niiden hajoamista.
Tämä uusi lääke on osoittanut tehoa myös potilailla, joilla LDL-reseptori ei toimi3,4.
Viime aikoina on myös yritetty ymmärtää, voisiko geeniterapia toimia, kun terve geeni lisätään potilaan DNA: han5.
Tästä luettelosta mahdollisista familiaalisen hyperkolesterolemian hoidoista on mahdollista ymmärtää, kuinka paljon tutkimusta tehdään ratkaisujen tarjoamiseksi näille potilaille, joita tämä vakava sairaus vaivaa harvinaisina, mikä vähentää kuolemaan johtavien seurausten riskiä.
Kuten uusien lääkkeiden kohdalla aina tapahtuu, on hyvä käyttää varovaisuutta niiden kroonisessa käytössä, koska tieto mahdollisista sivuvaikutuksista on selkeämpää vain kokemuksen (lääketurvatoiminnan) myötä, joten niiden käyttöä pediatriassa tulisi arvioida huolellisesti, tietysti lopullisena tavoitteena on tarjota näille "pienille" potilaille mahdollisuus elää "normaalia" elämää.
Perheen hyperkolesterolemiaa käsittelevän artikkelin bibliografiset ja sitografiset viitteet
1 2019 ESC / EAS-ohjeet dyslipidaemioiden hoitoon: Lipidien muokkaus kardiovaskulaarisen riskin vähentämiseksi. Kirjoittajat / työryhmän jäsenet; ESC: n käytäntöohjeiden komitea (CPG); ESC: n kansalliset sydänseurat. Ateroskleroosi. 2019 marraskuu; 290: 140-205. doi: 10.1016 / j.atherosclerosis.2019.08.014.
2 Harvinaiset dyslipidaemiat, fenotyypistä genotyypistä hallintaan: European Atherosclerosis Society -työryhmän yksimielinen lausuma. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet-diabetes endokrinoli. 2020 tammikuu; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoproteiini (a) Lipoproteiiniafereesin alentaminen antisense-oligonukleotidiin. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 3. heinäkuuta; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 LDL-kolesterolia alentava hoito. Pirillo A, Norata GD, Catapano AL.Handb Exp Pharmacol. 2020 30. huhtikuuta doi: 10.1007 / 164_2020_361.
5 Katsaus geeni- ja solupohjaisiin hoitoihin familiaaliseen hyperkolesterolemiaan. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A.Pharmacol Res. 2019 toukokuu; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.