
Hypercholestérolémie familiale: qu'est-ce que c'est et comment la traiter
Hypercholestérolémie familiale, une maladie génétique rare qui entraîne de graves conséquences sur la santé (par exemple, crise cardiaque et accident vasculaire cérébral) qui nécessite des mesures de confinement appropriées avec des médicaments appropriés et un mode de vie approprié.
Que signifie l'hypercholestérolémie? Et que signifie familial?
L'hypercholestérolémie est une affection clinique caractérisée par un cholestérol sanguin excessif.
Le cholestérol est un composant des lipides, généralement pris avec l'alimentation, mais qui peut également être produit par l'organisme.
Le cholestérol est très important pour la vie car il est utilisé pour la formation des membranes cellulaires (pour assurer leur fonction), protège les neurones et les nerfs crâniens.
Le cholestérol est également utilisé pour la synthèse d'autres molécules, dont trois sont les acides biliaires (importants pour la digestion), certaines hormones et la vitamine D.
Ce n'est que s'il est présent au-dessus de ces exigences qu'il peut causer de graves dommages.
Dans le cas de l'hypercholestérolémie, le cholestérol s'accumule dans le sang sous forme de lipoprotéines légères (LDL), des agrégats particuliers de graisses et de protéines, également appelés «mauvais cholestérol» qui favorisent la formation de plaques dans la paroi artérielle (plaques athérosclérotiques).
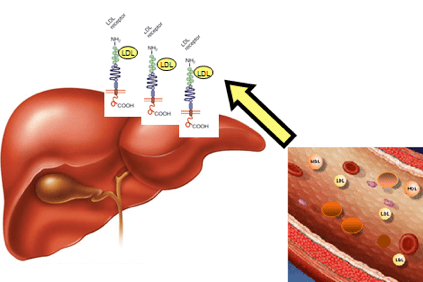
Cela se produit lorsque les patients ont des taux de cholestérol si élevés qu'ils ne peuvent pas l'éliminer par les mécanismes physiologiques du foie.
L'accumulation de LDL dans le sang conduit à terme à la formation de l'athérome (une obstruction physique à la circulation sanguine normale) qui peut entraîner des conséquences plus graves telles que l'angine, la crise cardiaque, l'accident vasculaire cérébral, mais aussi dans d'autres organes comme le cerveau, les reins, les poumons et le foie lui-même.
L'hypercholestérolémie est familière lorsqu'elle est transmise à la progéniture
Cela est dû aux modifications d'un gène qui contient les informations nécessaires pour fabriquer une protéine hépatique, le récepteur LDLR (LDLR), qui reconnaît le LDL, le supprime de la circulation sanguine et le transporte vers les cellules hépatiques, qui ensuite le suppriment. Cette altération du gène provoque une accumulation de LDL dans le sang.
À ce jour, il existe plus de 600 altérations connues du gène LDL qui provoquent une hypercholestérolémie familiale.
En outre, les personnes atteintes de ce trouble peuvent modifier d'autres gènes impliqués dans le métabolisme des lipoprotéines tels que le gène APOB (apoprotéine B dans LDL) et le gène PCSK9 (une protéine qui dégrade les récepteurs LDL).
Bien que l'on sache que l'accumulation de cholestérol peut être nocive, on estime que jusqu'à un tiers des crises cardiaques survenant avant l'âge de 40 ans sont dues à une hypercholestérolémie familiale non diagnostiquée ou traitée de manière inappropriée.
L'hypercholestérolémie multifactorielle causée par des facteurs environnementaux (une alimentation riche en graisses, surtout si elle est associée à une inactivité physique) est plus courante, même en présence de facteurs génétiques prédisposants.
Les formes dont se manifeste l'hypercholestérolémie familiale
Cette maladie peut se présenter sous 2 formes différentes: une moins grave (hétérozygote, 1 cas pour 500 individus) et une plus grave (homozygote, 1 cas pour 1,000,000 1,2 XNUMX d'individus) XNUMX.
La forme hétérozygote est souvent asymptomatique et n'est diagnostiquée que sur la base du taux de cholestérol sanguin.
Le foie a du mal à éliminer les LDL parce que les récepteurs LDL sont produits en nombre insuffisant, ce qui entraîne une augmentation de 2 ou 3 fois des taux sanguins par rapport aux valeurs normales.
Cette forme peut entraîner un risque accru de maladie cardiovasculaire à l'âge adulte.
La forme homozygote se caractérise par l'apparition de maladies cardiovasculaires même à un jeune âge et la présence d'accumulations caractéristiques de graisses telles que les xanthomes (nodules jaunâtres sur les articulations des mains et le tendon d'Achille) et le xanthélasma (plaques jaunâtres sur les paupières et autour les yeux).
Le défaut génétique est hérité des deux parents et, le risque de crise cardiaque en l'absence de traitement est déjà observé vers 15-20 ans.
En fait, dans cette condition, le foie ne parvient pas à métaboliser les lipoprotéines qui restent dans le sang, qui s'accumulent alors conduisant aux dysfonctionnements décrits ci-dessus et créent une situation incompatible avec la vie.
Quelles sont les chances de transmission de la maladie aux enfants?
Puisque chacun de nous a deux copies de chaque gène, chacune héritée d'un parent, nous aurons la forme hétérozygote lorsque nous hériterons d'une copie modifiée et d'une copie saine.
Au contraire, nous aurons la forme homozygote si nous héritons des deux copies du gène malade des deux parents. Il faut dire que cette forme est très rare car elle ne survient que lorsque les deux parents possèdent le gène qui cause la maladie et donc chacun d'eux peut transmettre une copie de la même altération génétique.
L'hypercholestérolémie familiale est appelée pathologie hétérozygote composée dans les rares cas où deux types différents d'altérations géniques sont hérités, un de chaque parent.
Les enfants à risque de développer la maladie doivent être diagnostiqués et traités tôt. Aujourd'hui, il est possible de diagnostiquer la maladie grâce à des tests génétiques qui recherchent des erreurs (altérations génétiques) sur les gènes LDLR, ApoB et PCSK9.
Par conséquent, seules les formes d'hypercholestérolémie familiale sont transmissibles.
Cependant, les formes multifactorielles qui surviennent principalement en raison d'un mode de vie incorrect (accumulation de cholestérol avec l'alimentation), peuvent également être caractérisées par une prédisposition génétique.
En fait, ces sujets peuvent présenter des déficits génétiques qui compromettent la capacité de l'organisme à compenser adéquatement l'excès de lipides dans l'alimentation.
Par exemple, lorsque le foie est saturé de cholestérol pris avec de la nourriture, la production de récepteurs qui capturent les LDL circulant dans le sang peut être supprimée.
Une situation est très similaire à celle qui survient lors d'une hypercholestérolémie familiale bien que moins sévère.
Dans ce cas, la concentration de cholestérol total dans le sang est supérieure à la normale et se situe généralement entre 240 et 350 mg / dl par rapport à la concentration normale fixée classiquement autour de 200 à 240 mg / dl.

Une curiosité: cette maladie peut avoir été transmise au cours des siècles
Une observation attentive de la célèbre peinture de la Joconde montre la possibilité que Mona Lisa déjà à un jeune âge souffre d'hypercholestérolémie familiale.
En fait, Leonardo décrit également fidèlement les dépôts graisseux typiques sur les mains et près des yeux (maintenant appelés xanthomes et xanthélasma) qui indiquent avec certitude la présence de la pathologie.
Si cela est vrai, il est démontré que l'hypercholestérolémie familiale est une maladie préservée au cours des siècles (un cas documenté dès 1500).
Quelles sont les possibilités de traiter cette maladie aujourd'hui?
La possibilité de guérir la maladie dépend de sa gravité. En outre, les facteurs de risque (régime alimentaire, tabagisme, âge, famille et antécédents personnels d'hypercholestérolémie, présence d'autres maladies) peuvent aggraver le tableau général de la maladie.
Pour traiter l'hypercholestérolémie familiale, des médicaments de nouvelle génération peuvent être utilisés, mais en même temps, il est nécessaire d'agir avec une modification appropriée du mode de vie.
En cas de gravité intermédiaire (hypercholestérolémie de la famille hétérozygote), un traitement médicamenteux à base de statine (inhibiteurs de la synthèse du cholestérol qui induisent une augmentation de la synthèse des récepteurs LDLR) et l'association avec l'ézétimibe (inhibiteurs de l'absorption du cholestérol) ou les inhibiteurs de PCSK9 (c'est-à-dire les inhibiteurs de détruit les récepteurs hépatiques qui capturent le LDLR) améliore l'activité de la copie saine du gène LDLR et réduit l'accumulation de cholestérol dans le sang.
Récemment approuvé est également l'acide bempédoïque, un médicament qui agit dans le foie en inhibant l'enzyme ATP citrate lyase, une molécule impliquée dans le processus de synthèse endogène du cholestérol.
Ce mécanisme d'action nous permet d'agir sur la quantité de cholestérol produit, agissant en amont du site d'action des statines, et de stimuler l'expression des récepteurs LDL pour compenser la synthèse réduite.
Contrairement aux statines, l'acide bempédoïque n'est pas actif dans le muscle squelettique, ce qui diminue la possibilité d'avoir des événements indésirables typiques des statines.
Ce médicament peut également être associé à l'ézétimibe avec des effets favorables.
L'hypercholestérolémie familiale homozygote était jusqu'à récemment considérée comme une maladie incurable.
La thérapie à base de statines n'est pas efficace dans cette pathologie. En effet, les statines qui agissent sur les mécanismes conduisant à la production de cholestérol endogène ne peuvent pas stimuler la synthèse des récepteurs LDL.
Pour éliminer le cholestérol LDL de l'organisme de ces patients, la plasmaphérèse, une technique qui permet de filtrer le sang en éliminant les graisses, à l'instar de ce qui se fait avec la dialyse lorsque les reins ne fonctionnent pas.
Cependant, il s'agit d'une procédure invasive ayant un impact négatif sur la qualité de vie des patients.
Les dernières recherches sur l'hypercholestérolémie familiale
Ces dernières années, la recherche a conduit au développement de médicaments spécifiques également pour la forme homozygote de l'hypercholestérolémie familiale, améliorant considérablement l'espérance et la qualité de vie des patients qui en souffrent.
Par exemple, le médicament lomitapide (pris par voie orale) conduit à des taux plasmatiques de LDL-cholestérol nettement inférieurs chez ces patients.
Le lomitapide inhibe une protéine microsomale de transport des triglycérides (MTP) qui permet l'incorporation de ces lipides avec l'apoprotéine B100 dans les VLDL naissantes.
En conséquence, l'apoprotéine B100 et le LDL sont réduits même chez les patients où la maladie a provoqué l'absence totale du récepteur LDL.
Un autre médicament de nouvelle génération est le mipomersen, un oligonucléotide antisens capable de dégrader les apoprotéines B100 qui participent à la formation des LDL, réduisant ainsi le nombre de ces derniers.
L'évolocumab et l'alirocumab sont deux anticorps monoclonaux qui inhibent l'activité de la protéine PCSK9 dans le sang.
Cependant, pour que ces deux médicaments prennent effet, au moins une petite partie des récepteurs LDL doit être présente et fonctionner.
Parmi les médicaments encore en développement clinique, il y a l'inclusion d'un médicament biologique (appelé siRNA) qui bloque la transcription de l'ADN et la synthèse de la protéine PCSK9 dans le foie.
Des résultats positifs ont été récemment obtenus avec l'évinacumab, un anticorps monoclonal inhibiteur de la protéine similaire-angiopoïétine 3 (ANGPTL3), une molécule synthétisée par le foie dont le rôle est d'augmenter les taux de cholestérol-LLDL et de triglycérides empêchant leur dégradation.
Ce nouveau médicament a également montré son efficacité chez les patients dont le récepteur LDL ne fonctionne pas3,4.
Récemment, des tentatives ont également été faites pour comprendre si la thérapie génique, avec l'introduction du gène sain dans l'ADN du patient, pouvait fonctionner5.
À partir de cette liste de thérapies possibles pour l'hypercholestérolémie familiale, il est possible de comprendre combien de recherches sont menées pour apporter des solutions à ces patients qui, bien que rares, sont atteints de cette maladie grave, réduisant ainsi le risque de conséquences mortelles.
Cependant, comme toujours avec les nouveaux médicaments, il est bon de mettre en garde contre leur utilisation chronique car la connaissance de leurs effets secondaires possibles ne sera plus claire qu'avec l'expérience (pharmacovigilance), leur utilisation en pédiatrie doit donc être soigneusement évaluée, bien sûr dans le but ultime d'offrir à ces «petits» patients la possibilité de mener une vie «normale».
Références bibliographiques et sitographiques pour l'article sur l'hypercholestérolémie familiale
1 Lignes directrices 2019 ESC / EAS pour la prise en charge des dyslipidémies: modification lipidique pour réduire le risque cardiovasculaire. Auteurs / membres du groupe de travail; Comité ESC pour les directives pratiques (CPG); Sociétés nationales de cardiologie ESC. Athérosclérose. 2019 novembre; 290: 140-205. doi: 10.1016 / j.atherosclerosis.2019.08.014.
2 Dyslipidémies rares, du phénotype au génotype en passant par la prise en charge: déclaration de consensus du groupe de travail de l'European Atherosclerosis Society. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet Diabetes Endocrinol. 2020 janvier; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoprotéine (a) Abaissement de l'aphérèse des lipoprotéines à l'approche oligonucléotidique antisens. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 juillet 3; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 Thérapie abaissant le cholestérol LDL. Pirillo A, Norata GD, Catapano AL.Handb Exp Pharmacol. 2020 avril 30 doi: 10.1007 / 164_2020_361.
5 Une revue des thérapies génétiques et cellulaires pour l'hypercholestérolémie familiale. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner®, Sahebkar A.Pharmacol Res. 2019 mai; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.