
Familiehypercholesterolemie: wat het is en hoe het te behandelen
Familiaire hypercholesterolemie, een zeldzame genetische ziekte die ernstige gevolgen voor de gezondheid heeft (bijv. Hartaanval en beroerte) waarvoor passende inperkingsmaatregelen met geschikte medicatie en een passende levensstijl nodig zijn.
Wat betekent hypercholesterolemie? En wat betekent familiair?
Hypercholesterolemie is een klinische aandoening die wordt gekenmerkt door een te hoog cholesterolgehalte in het bloed.
Cholesterol is een bestanddeel van lipiden, dat meestal met de voeding wordt ingenomen, maar dat ook door het lichaam kan worden aangemaakt.
Cholesterol is erg belangrijk voor het leven omdat het wordt gebruikt voor de vorming van celmembranen (om hun functie te waarborgen), neuronen en hersenzenuwen beschermt.
Cholesterol wordt ook gebruikt voor de synthese van andere moleculen, waarvan er drie galzuren zijn (belangrijk voor de spijsvertering), sommige hormonen en vitamine D.
Alleen indien aanwezig boven deze vereisten kan het ernstige schade veroorzaken.
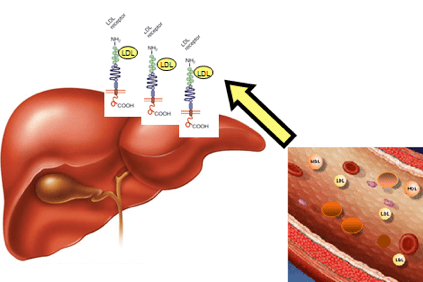
In het geval van hypercholesterolemie hoopt cholesterol zich op in het bloed in de vorm van lichte lipoproteïnen (LDL), bepaalde aggregaten van vetten en eiwitten, ook wel "slechte cholesterol" genoemd, die de vorming van plaques in de aderwand bevorderen (atherosclerotische plaques).
Dit gebeurt wanneer patiënten zo'n hoog cholesterolgehalte hebben dat ze het niet kunnen elimineren via de fysiologische mechanismen van de lever.
De ophoping van LDL in het bloed leidt uiteindelijk tot de vorming van het atheroma (een fysieke obstructie van de normale bloedstroom) wat kan leiden tot ernstigere gevolgen zoals angina pectoris, hartaanval, beroerte, maar ook in andere organen zoals de hersenen, nieren, longen en de lever zelf.
Hypercholesterolemie is bekend bij overdracht op nakomelingen
Dit komt door modificaties van een gen dat de informatie bevat om een levereiwit te maken, de LDLR (LDLR) -receptor, die LDL herkent, het uit de bloedbaan verwijdert en naar de levercellen transporteert, die het vervolgens weer verwijderen. Deze genverandering veroorzaakt LDL-accumulatie in het bloed.
Tot op heden zijn er meer dan 600 bekende veranderingen in het LDL-gen die familiehypercholesterolemie veroorzaken.
Mensen met deze aandoening kunnen ook veranderingen aanbrengen in andere genen die betrokken zijn bij het metabolisme van lipoproteïnen, zoals het APOB-gen (apoproteïne B in LDL) en het PCSK9-gen (een eiwit dat LDL-receptoren afbreekt).
Hoewel bekend is dat de accumulatie van cholesterol schadelijk kan zijn, wordt geschat dat tot een derde van de hartaanvallen die vóór de leeftijd van 40 plaatsvinden, te wijten is aan niet-gediagnosticeerde of onjuist behandelde hypercholesterolemie in het gezin.
Vaker komt multifactoriële hypercholesterolemie voor die wordt veroorzaakt door omgevingsfactoren (een dieet met veel vet, vooral als dit gepaard gaat met lichamelijke inactiviteit), zelfs in de aanwezigheid van predisponerende genetische factoren.
De vormen waarin familiehypercholesterolemie zich manifesteert
Deze ziekte kan zich in 2 verschillende vormen voordoen: een minder ernstige (heterozygoot, 1 geval per 500 individuen) en een ernstiger (homozygoot, 1 geval per 1,000,000 individuen) 1,2.
De heterozygote vorm is vaak asymptomatisch en wordt alleen gediagnosticeerd op basis van het cholesterolgehalte in het bloed.
De lever heeft moeite om LDL te elimineren omdat LDL-receptoren in onvoldoende aantallen worden geproduceerd, wat resulteert in een twee- of drievoudige toename van de bloedspiegels in vergelijking met normale waarden.
Deze vorm kan op volwassen leeftijd leiden tot een verhoogd risico op hart- en vaatziekten.
De homozygote vorm wordt gekenmerkt door het begin van hart- en vaatziekten zelfs op jonge leeftijd en de aanwezigheid van karakteristieke vetophopingen zoals xanthomas (gelige knobbeltjes op de knokkels van de handen en de achillespees) en xanthelasma (gelige platen op de oogleden en rond de ogen).
Het genetisch defect wordt overgeërfd van beide ouders en het risico op een hartaanval zonder therapie wordt al waargenomen rond de leeftijd van 15-20 jaar.
In feite slaagt de lever er in deze toestand niet in om de lipoproteïnen die in het bloed achterblijven te metaboliseren, die zich vervolgens ophopen, wat leidt tot de hierboven beschreven disfuncties en een situatie creëren die onverenigbaar is met het leven.
Hoe groot is de kans dat de ziekte op kinderen wordt overgedragen?
Aangezien ieder van ons twee kopieën van elk gen heeft, elk geërfd van een ouder, zullen we de heterozygote vorm hebben als we één gewijzigde en één gezonde kopie erven.
Integendeel, we zullen de homozygote vorm hebben als we beide kopieën van het zieke gen van beide ouders erven. Het moet gezegd worden dat deze vorm zeer zeldzaam is, omdat het alleen voorkomt als beide ouders het gen hebben dat de ziekte veroorzaakt en daarom elk van hen een kopie van dezelfde genwijziging kan doorgeven.
Familiaire hypercholesterolemie wordt heterozygote pathologie genoemd en bestaat in de zeldzame gevallen waarin twee verschillende soorten genveranderingen worden overgeërfd, één van elke ouder.
Kinderen die het risico lopen de ziekte te ontwikkelen, moeten vroegtijdig worden gediagnosticeerd en behandeld. Tegenwoordig is het mogelijk om de ziekte te diagnosticeren door middel van genetische tests die zoeken naar fouten (genveranderingen) op LDLR-, ApoB- en PCSK9-genen.
Daarom zijn alleen de vormen van familiehypercholesterolemie overdraagbaar.
Echter, multifactoriële vormen die voornamelijk ontstaan door een verkeerde leefstijl (ophoping van cholesterol met de voeding), kunnen ook gekenmerkt worden door een erfelijke aanleg.
In feite kunnen deze personen genetische tekortkomingen hebben die het vermogen van het lichaam om overtollige lipiden in de voeding adequaat te compenseren, in gevaar brengen.
Als de lever bijvoorbeeld verzadigd is met cholesterol dat met voedsel wordt ingenomen, kan de productie van receptoren die LDL-circulatie in het bloed opvangen, worden onderdrukt.
Een situatie lijkt sterk op de situatie die optreedt tijdens hypercholesterolemie in het gezin, hoewel minder ernstig.
In dit geval is de concentratie van totaal cholesterol in het bloed hoger dan normaal en ligt deze gewoonlijk tussen 240 en 350 mg / dl in vergelijking met de normale concentratie die gewoonlijk wordt vastgesteld op 200 - 240 mg / dl.

Een curiosum: deze ziekte is mogelijk door de eeuwen heen overgedragen
Een zorgvuldige observatie van het beroemde schilderij van de Mona Lisa toont de mogelijkheid aan dat Mona Lisa al op jonge leeftijd leed aan hypercholesterolemie in de familie.
In feite geeft Leonardo ook getrouw de typische vetophopingen op de handen en nabij de ogen weer (nu bekend als xanthomas en xanthelasma) die de aanwezigheid van de pathologie met zekerheid aangeven.
Als dit waar is, wordt aangetoond dat hypercholesterolemie in de familie een ziekte is die door de eeuwen heen is behouden (een geval dat al in 1500 werd gedocumenteerd).
Wat zijn de mogelijkheden om deze ziekte vandaag te behandelen?
De mogelijkheid om de ziekte te genezen, hangt af van de ernst ervan. Ook kunnen risicofactoren (dieet, roken, leeftijd, familie en persoonlijke geschiedenis van hypercholesterolemie, aanwezigheid van andere ziekten) het algemene ziektebeeld verslechteren.
Om familiehypercholesterolemie te behandelen, kunnen nieuwe generatie medicijnen worden gebruikt, maar tegelijkertijd is het noodzakelijk om te handelen met een geschikte aanpassing van de levensstijl.
In gevallen van middelmatige ernst (heterozygote familie hypercholesterolemie), statine-gebaseerde medicamenteuze therapie (cholesterolsyntheseremmers die een verhoogde LDLR-receptorsynthese induceren) en de combinatie met ezetimibe (cholesterolabsorptieremmers) of PCSK9-remmende geneesmiddelen (dwz PCSK9-eiwitremmers waarvan de rol is leverreceptoren vernietigen die LDLR opvangen) verbetert de activiteit van de gezonde kopie van het LDLR-gen en vermindert de ophoping van cholesterol in het bloed.
Onlangs goedgekeurd is ook bempedoïnezuur, een medicijn dat in de lever werkt door het enzym ATP-citraatlyase te remmen, een molecuul dat betrokken is bij het proces van endogene cholesterolsynthese.
Dit werkingsmechanisme stelt ons in staat om in te werken op de hoeveelheid geproduceerde cholesterol, stroomopwaarts van de plaats van werking van de statines, en om de expressie van LDL-receptoren te stimuleren om de verminderde synthese te compenseren.
In tegenstelling tot statines is bempedoïnezuur niet actief in skeletspieren, waardoor de kans op ongewenste gebeurtenissen die kenmerkend zijn voor statines, kleiner wordt.
Dit medicijn kan ook worden geassocieerd met ezetimibe met gunstige effecten.
Homozygote familiaire hypercholesterolemie werd tot voor kort als een ongeneeslijke ziekte beschouwd.
Op statines gebaseerde therapie is niet effectief bij deze pathologie. Statines die werken op de mechanismen die leiden tot de productie van endogeen cholesterol, kunnen de synthese van LDL-receptoren niet stimuleren.
Om LDL-cholesterol uit het lichaam van deze patiënten te verwijderen, plasmaferese, een techniek die het mogelijk maakt om het bloed te filteren door vetten te verwijderen, net zoals bij dialyse wanneer de nieren niet werken.
Dit is echter een invasieve procedure met een negatieve invloed op de kwaliteit van leven van patiënten.
Het laatste onderzoek naar hypercholesterolemie in het gezin
In de afgelopen jaren heeft onderzoek geleid tot de ontwikkeling van specifieke geneesmiddelen, ook voor de homozygote vorm van hypercholesterolemie in het gezin, waardoor de verwachting en de kwaliteit van leven van patiënten die eraan lijden aanzienlijk zijn verbeterd.
Het medicijn lomitapide (oraal ingenomen) leidt bijvoorbeeld tot duidelijk lagere plasma LDL-cholesterolwaarden bij deze patiënten.
Lomitapide remt een microsomaal triglyceridetransporteiwit (MTP) dat de opname van deze lipiden samen met het apoproteïne B100 in opkomende VLDL's mogelijk maakt.
Als resultaat worden de apoproteïne B100 en LDL zelfs verminderd bij patiënten bij wie de ziekte de totale afwezigheid van de LDL-receptor heeft veroorzaakt.
Een ander geneesmiddel van de nieuwe generatie is mipomersen, een antisense-oligonucleotide dat in staat is de B100-apoproteïnen die deelnemen aan de vorming van LDL, af te breken, waardoor het aantal van de laatste wordt verminderd.
Evolocumab en alirocumab zijn twee monoklonale antilichamen die de activiteit van het PCSK9-eiwit in het bloed remmen.
Om deze twee geneesmiddelen echter te laten werken, moet ten minste een klein deel van de LDL-receptoren aanwezig zijn en functioneren.
Een van de geneesmiddelen die nog in klinische ontwikkeling zijn, is de opname van een biologisch geneesmiddel (het zogenaamde siRNA) dat de DNA-transcriptie en synthese van het PCSK9-eiwit in de lever blokkeert.
Positieve resultaten zijn onlangs bereikt met evinacumab, een monoklonale antilichaamremmer van het proteïne soortgelijk angiopoëtine 3 (ANGPTL3), een door de lever gesynthetiseerd molecuul met als rol het verhogen van de cholesterol-LLDL- en triglycerideniveaus om hun afbraak te voorkomen.
Dit nieuwe medicijn is ook effectief gebleken bij patiënten bij wie de LDL-receptor niet functioneert3,4.
Onlangs zijn er ook pogingen gedaan om te begrijpen of gentherapie, met de introductie van het gezonde gen in het DNA van de patiënt, zou kunnen werken5.
Uit deze lijst met mogelijke therapieën voor familiaire hypercholesterolemie is het mogelijk om te begrijpen hoeveel onderzoek er wordt gedaan om oplossingen te bieden voor deze patiënten die, hoewel zeldzaam, aan deze ernstige ziekte lijden, waardoor het risico op fatale gevolgen wordt verminderd.
Zoals altijd gebeurt met nieuwe geneesmiddelen, is het echter goed om een woord van waarschuwing te gebruiken over hun chronisch gebruik, aangezien de kennis over hun mogelijke bijwerkingen alleen duidelijker zal zijn met ervaring (geneesmiddelenbewaking), dus hun gebruik in de kindergeneeskunde moet zorgvuldig worden geëvalueerd, uiteraard met het uiteindelijke doel om deze “kleine” patiënten de mogelijkheid te bieden een “normaal” leven te leiden.
Bibliografische en sitografische verwijzingen voor het artikel over hypercholesterolemie in het gezin
1 2019 ESC / EAS-richtlijnen voor het beheer van dyslipidemieën: lipidenmodificatie om cardiovasculair risico te verminderen. Auteurs / leden van de Task Force; ESC-commissie voor praktijkrichtlijnen (CPG); ESC National Cardiac Societies. Atherosclerose. 2019 november; 290: 140-205. doi: 10.1016 / j.atherosclerose.2019.08.014.
2 Zeldzame dyslipidemieën, van fenotype tot genotype tot management: een consensusverklaring van een taskforce van de European Atherosclerosis Society. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet Diabetes Endocrinol. 2020 januari; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoproteïne (a) Verlaging van lipoproteïne-aferese naar antisense-oligonucleotide-benadering. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 juli 3; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 LDL-cholesterolverlagende therapie. Pirillo A, Norata GD, Catapano AL.Handb Exp Pharmacol. 2020 april 30 doi: 10.1007 / 164_2020_361.
5 Een overzicht van gen- en celgebaseerde therapieën voor familiaire hypercholesterolemie. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A.Pharmacol Res. 2019 mei; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.