
Perekonna hüperkolesteroleemia: mis see on ja kuidas seda ravida
Perekondlik hüperkolesteroleemia, haruldane geneetiline haigus, mis põhjustab tõsiseid tervisekahjustusi (nt südameatakk ja insult), mis nõuab asjakohaseid ohjeldamismeetmeid koos sobivate ravimite ja sobiva eluviisiga.
Mida tähendab hüperkolesteroleemia? Ja mida tähendab perekondlik?
Hüperkolesteroleemia on kliiniline seisund, mida iseloomustab liigne kolesteroolisisaldus veres.
Kolesterool on lipiidide komponent, mida võetakse tavaliselt koos toiduga, kuid mida võib toota ka keha.
Kolesterool on kogu elu jaoks väga oluline, kuna seda kasutatakse rakumembraanide moodustamiseks (nende funktsiooni tagamiseks), kaitseb neuroneid ja kraniaalnärve.
Kolesterooli kasutatakse ka teiste molekulide sünteesiks, millest kolm on sapphapped (seedimiseks olulised), mõned hormoonid ja D-vitamiin.
Ainult siis, kui need on kõrgemad kui need nõuded, võib see põhjustada tõsist kahju.
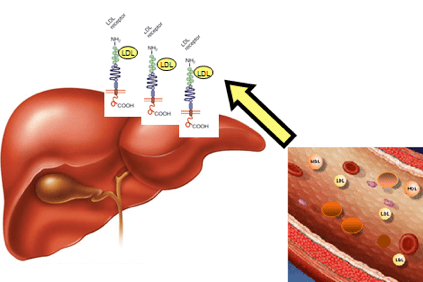
Hüperkolesteroleemia korral koguneb kolesterool veres kergete lipoproteiinide (LDL), eriti rasvade ja valkude kogumite kujul, mida nimetatakse ka „halvaks kolesterooliks“ ja mis soodustavad naastude moodustumist arteri seinas (aterosklerootilised naastud).
See juhtub siis, kui patsientidel on nii kõrge kolesteroolitase, et nad ei suuda seda maksa füsioloogiliste mehhanismide kaudu kõrvaldada.
LDL-i akumuleerumine veres viib lõpuks ateroomi (normaalse verevoolu füüsilise takistuseni) moodustumiseni, mis võib viia tõsisemate tagajärgedeni nagu stenokardia, südameatakk, insult, aga ka teistes elundites, näiteks ajus, neerud, kopsud ja maks ise.
Hüperkolesteroleemia on järglastele levinud tuttav
Selle põhjuseks on maksa valgu valmistamiseks vajalikku teavet sisaldava geeni modifikatsioonid - LDLR (LDLR) retseptor, mis tunneb ära LDL, eemaldab selle vereringest ja transpordib maksarakkudesse, mis seejärel eemaldavad. See geenimuutus põhjustab LDL-i akumuleerumist veres.
Praeguseks on LDL-geenis teada üle 600 muutuse, mis põhjustavad perekonna hüperkolesteroleemiat.
Samuti võivad selle häirega inimesed põhjustada muutusi teistes lipoproteiini metabolismis osalevates geenides, näiteks APOB-geen (apoproteiin B LDL-is) ja PCSK9-geen (valk, mis lagundab LDL-retseptoreid).
Ehkki on teada, et kolesterooli akumuleerumine võib olla kahjulik, on hinnanguliselt kuni kolmandik enne 40. eluaastat tekkinud südameatakkidest tingitud diagnoosimata või valesti ravitud perekonna hüperkolesteroleemiast.
Sagedasem on keskkonnateguritest põhjustatud multifaktoriaalne hüperkolesteroleemia (kõrge rasvasisaldusega dieet, eriti kui see on seotud füüsilise passiivsusega) isegi eelsooduvate geneetiliste tegurite olemasolul.
Vormid, mis perekonna hüperkolesteroleemia avalduvad
See haigus võib avalduda kahel erineval kujul: vähem tõsine (heterosügootne, 2 juhtum iga 1 inimese kohta) ja raskem (homosügootne, 500 juhtum iga 1 1,000,000 1,2 inimese kohta) XNUMX.
Heterosügootne vorm on sageli asümptomaatiline ja diagnoositakse ainult vere kolesteroolitaseme põhjal.
Maks võitleb LDL-i kõrvaldamisega, kuna LDL-retseptoreid toodetakse ebapiisavas koguses, mille tulemuseks on vere normaalse taseme tõus 2 või 3 korda.
See vorm võib täiskasvanueas suurendada südame-veresoonkonna haiguste riski.
Homosügootset vormi iseloomustab kardiovaskulaarsete haiguste ilmnemine isegi noores eas ning iseloomulike rasvade kogunemine nagu ksantoomid (kollakad sõlmed käte ja Achilleuse kõõluse sõrmenukkidel) ja ksanthelasma (kollakad plaadid silmalaugudel ja selle ümbruses) silmad).
Geneetiline defekt on päritud mõlemalt vanemalt ja südameataki riski teraapia puudumisel täheldatakse juba umbes 15-20-aastaselt.
Tegelikult ei suuda maks selles seisundis metaboliseerida veres püsivaid lipoproteiine, mis seejärel kogunevad, põhjustades ülalkirjeldatud talitlushäireid ja tekitades eluga kokkusobimatu olukorra.
Kui suur on haiguse edasikandumise tõenäosus lastele?
Kuna meil kõigil on igast geenist kaks eksemplari, mis mõlemad on päritud vanemalt, on meil heterosügootne vorm, kui pärime ühe muudetud ja ühe terve koopia.
Vastupidi, meil on homosügootne vorm, kui pärime haigestunud geeni mõlemad koopiad mõlemalt vanemalt. Tuleb öelda, et see vorm on väga haruldane, kuna see esineb ainult siis, kui mõlemal vanemal on haigus põhjustav geen ja seetõttu saavad kõik neist edastada sama geenimuutuse koopia.
Perekondlikku hüperkolesteroleemiat nimetatakse heterosügootseks patoloogiaks, mis koosneb harvadel juhtudel, kui päritakse kaks erinevat tüüpi geenimuutusi, üks mõlemalt vanemalt.
Lapsed, kellel on risk haigestuda, tuleb varakult diagnoosida ja ravida. Täna on haigust võimalik diagnoosida geneetiliste testide abil, mis otsivad vigu (geenimuutusi) LDLR, ApoB ja PCSK9 geenides.
Seetõttu on ülekantavad ainult perekonna hüperkolesteroleemia vormid.
Kuid mitmefaktorilisi vorme, mis tekivad peamiselt vale eluviisi tõttu (kolesterooli kogunemine dieediga), võib iseloomustada ka geneetilise eelsoodumusega.
Tegelikult võivad nendel subjektidel esineda geneetilised puudujäägid, mis kahjustavad keha võimet toidus sisalduva lipiidide liigset piisavat kompenseerimist.
Näiteks kui maks on küllastunud toiduga võetud kolesterooliga, võib retseptorite tootmine, mis haaravad veres ringlevat LDL-i, pärssida.
Olukord on väga sarnane olukorraga, mis esineb perekonna hüperkolesteroleemia korral, kuigi see on vähem tõsine.
Sellisel juhul on üldkolesterooli kontsentratsioon veres üle normi ja on tavaliselt vahemikus 240–350 mg / dl, võrreldes tavapärasega, mis on tavaliselt seatud umbes 200–240 mg / dl.

Uudishimu: see haigus võis olla sajandite vältel üle antud
Mona Lisa kuulsa maali hoolikas jälgimine näitab võimalust, et Mona Lisa kannatas juba noorena pere hüperkolesteroleemia all.
Tegelikult kujutab Leonardo tõetruult ka tüüpilisi kätes ja silmade läheduses asuvaid rasvaladestusi (nüüd tuntud kui ksantoomid ja ksanthelasma), mis viitavad kindlalt patoloogia olemasolule.
Kui see on tõsi, näidatakse, et perekonna hüperkolesteroleemia on haigus, mis on säilinud sajandite jooksul (juhtum on dokumenteeritud juba 1500. aastal).
Millised on selle haiguse ravimise võimalused tänapäeval?
Haiguse ravimise võimalus sõltub selle tõsidusest. Samuti võivad riskifaktorid (toitumine, suitsetamine, vanus, perekond ja hüperkolesteroleemia isiklik ajalugu, teiste haiguste esinemine) halvendada üldist haiguspilti.
Perekonna hüperkolesteroleemia raviks võib kasutada uue põlvkonna ravimeid, kuid samal ajal on vaja tegutseda elustiili asjakohase muutmisega.
Keskmise raskusastmega (heterosügootse perekonna hüperkolesteroleemia) korral kasutatakse statiinil põhinevat ravimit (kolesterooli sünteesi inhibiitorid, mis indutseerivad LDLR retseptori suurenenud sünteesi) ja kombinatsioonis esetimiibiga (kolesterooli imendumise inhibiitorid) või PCSK9 inhibeerivate ravimitega (st PCSK9 valgu inhibiitoritega, kelle roll on hävitavad maksa retseptorid, mis haaravad LDLR-i) parandab LDLR-geeni terve koopia aktiivsust ja vähendab kolesterooli kogunemist veres.
Hiljuti heaks kiidetud ravim on ka bempedoehape, ravim, mis toimib maksas, pärssides ensüümi ATP tsitraatlüaasi - molekuli, mis osaleb endogeense kolesterooli sünteesi protsessis.
See toimemehhanism võimaldab meil toimida toodetud kolesterooli kogusele, toimides statiinide toimekohast ülesvoolu, ja stimuleerida LDL-retseptorite ekspressiooni, et kompenseerida vähenenud sünteesi.
Erinevalt statiinidest ei ole bempedoehape skeletilihastes aktiivne, mis vähendab statiinidele omaste soovimatute sündmuste tekkimise võimalust.
Seda ravimit võib seostada ka soodsa toimega esetimiibiga.
Homosügootset perekondlikku hüperkolesteroleemiat peeti kuni viimase ajani ravimatu haigusena.
Statiinipõhine ravi ei ole selle patoloogia korral efektiivne. Tegelikult ei saa statiinid, mis toimivad mehhanismides, mis põhjustavad endogeense kolesterooli tootmist, stimuleerida LDL-retseptorite sünteesi.
LDL-kolesterooli väljutamiseks nende patsientide kehast plasmaferees, meetod, mis võimaldab verd filtreerida rasvade eemaldamise teel, sarnaselt dialüüsiga tehtavale, kui neerud ei tööta.
See on aga invasiivne protseduur, millel on negatiivne mõju patsientide elukvaliteedile.
Perekonna hüperkolesteroleemia uusimad uuringud
Viimastel aastatel on uuringud viinud spetsiifiliste ravimite väljatöötamiseni ka perekonna hüperkolesteroleemia homosügootse vormi jaoks, parandades oluliselt selle all kannatavate patsientide ootusi ja elukvaliteeti.
Näiteks põhjustab ravim lomitapiid (suu kaudu manustatuna) nendel patsientidel märgatavalt madalamat LDL-kolesterooli taset plasmas.
Lomitapiid pärsib mikrosomaalset triglütseriidide transportvalku (MTP), mis võimaldab neid lipiide koos apoproteiiniga B100 inkorporeerida tekkivatesse VLDL-idesse.
Selle tulemusel vähenevad apoproteiin B100 ja LDL isegi patsientidel, kelle haigus on põhjustanud LDL-retseptori täieliku puudumise.
Teine uue põlvkonna ravim on mipomerseen, antisenss-oligonukleotiid, mis on võimeline lagundama B100 apoproteiine, mis osalevad LDL-i moodustumisel, vähendades seeläbi viimaste arvu.
Evolokumab ja alirokumab on kaks monoklonaalset antikeha, mis pärsivad PCSK9 valgu aktiivsust veres.
Kuid nende kahe ravimi toimimiseks peab kohal olema ja toimima vähemalt väike osa LDL-retseptoritest.
Jätkuvalt kliinilises arengus olevate ravimite hulka kuulub bioloogilise ravimi (nn siRNA) lisamine, mis blokeerib maksa transkriptsiooni ja PCSK9 valgu sünteesi maksas.
Hiljuti on saavutatud positiivseid tulemusi evinakumabiga, mis on valgu analoog-angiopoietiin 3 (ANGPTL3) monoklonaalse antikeha inhibiitor - maksa sünteesitud molekul, mille ülesanne on suurendada kolesterooli-LLDL ja triglütseriidide taset, vältides nende lagunemist.
See uus ravim on näidanud efektiivsust ka patsientidel, kellel LDL-retseptor ei tööta3,4.
Hiljuti on püütud mõista ka seda, kas geeniteraapia koos terve geeni viimisega patsiendi DNA-sse võiks toimida5.
Sellest perekondliku hüperkolesteroleemia võimalike ravimeetodite loendist on võimalik mõista, kui palju uuritakse, et pakkuda lahendusi nendele patsientidele, keda see raske haigus vaevab, ehkki harva, vähendades surmaga lõppevate tagajärgede riski.
Kuid nagu uute ravimite puhul alati juhtub, on nende kroonilisel kasutamisel hea kasutada ettevaatusega sõna, kuna teadmised nende võimalike kõrvaltoimete kohta on selgemad ainult kogemuste (ravimiohutuse järelevalve) korral, mistõttu tuleks nende kasutamist pediaatrias hoolikalt hinnata. muidugi lõppeesmärgiga pakkuda neile “väikestele” patsientidele võimalust elada “normaalset” elu.
Perekonna hüperkolesteroleemiat käsitleva artikli bibliograafilised ja sitograafilised viited
1 2019 ESC / EAS suunised düslipideemiate raviks: lipiidide muutmine kardiovaskulaarse riski vähendamiseks. Autorid / rakkerühma liikmed; ESC praktikajuhiste komitee (CPG); ESC riiklikud südameühingud. Ateroskleroos. 2019 november; 290: 140-205. doi: 10.1016 / j.atherosclerosis.2019.08.014.
2 Harvad düslipideemiad fenotüübist genotüübini juhtimiseni: Euroopa Ateroskleroosi Seltsi töörühma konsensuse avaldus. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lanceti diabeet endokrinool. 2020 jaanuar; 8 (1): 50–67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoproteiin (a) lipoproteiinide afereesi alandamine antisense oligonukleotiidide meetodile. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 3. juuli; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 LDL-kolesterooli alandav ravi. Pirillo A, Norata GD, Catapano AL. Handb Exp Pharmacol. 2020 30. aprill doi: 10.1007 / 164_2020_361.
5 Ülevaade perekondliku hüperkolesteroleemia geeni- ja rakupõhistest ravimeetoditest. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A. Pharmacol Res. 2019 mai; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.