
家族の高コレステロール血症:それは何であり、それをどのように治療するか
家族性高コレステロール血症は、深刻な健康への影響(心臓発作や脳卒中など)を引き起こすまれな遺伝性疾患であり、適切な投薬と適切なライフスタイルによる適切な封じ込め措置が必要です。
高コレステロール血症とはどういう意味ですか? そして、家族はどういう意味ですか?
高コレステロール血症は、血中の過剰なコレステロールを特徴とする臨床症状です。
コレステロールは脂質の成分であり、通常は食事と一緒に摂取されますが、体内で生成されることもあります。
コレステロールは、細胞膜の形成(機能を確保するため)に使用され、ニューロンと脳神経を保護するため、生命にとって非常に重要です。
コレステロールは他の分子の合成にも使用され、そのうちのXNUMXつは胆汁酸(消化に重要)、いくつかのホルモン、およびビタミンDです。
これらの要件を超えて存在する場合にのみ、重大な損傷を引き起こす可能性があります。
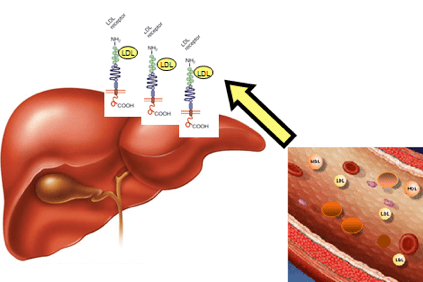
高コレステロール血症の場合、コレステロールは、軽いリポタンパク質(LDL)、動脈壁のプラーク(アテローム性動脈硬化症のプラーク)の形成を促進する「悪玉コレステロール」とも呼ばれる脂肪とタンパク質の特定の凝集体の形で血中に蓄積します。
これは、患者のコレステロール値が非常に高く、肝臓の生理学的メカニズムによってコレステロール値を除去できない場合に発生します。
血液中のLDLの蓄積は、最終的にアテローム(正常な血流の物理的閉塞)の形成につながり、狭心症、心臓発作、脳卒中などのより深刻な結果につながる可能性がありますが、脳などの他の臓器にも起こります。腎臓、肺、肝臓自体。
高コレステロール血症は、子孫に伝染するとよく知られています
これは、肝臓タンパク質を作るための情報を含む遺伝子の修飾によるものです。LDLR(LDLR)受容体は、LDLを認識し、血流から取り除き、肝臓細胞に輸送して、肝臓細胞から取り除きます。 この遺伝子の変化は、血中のLDL蓄積を引き起こします。
現在までに、家族の高コレステロール血症を引き起こすLDL遺伝子の600以上の既知の変化があります。
また、この障害を持つ人々は、APOB遺伝子(LDLのアポタンパク質B)やPCSK9遺伝子(LDL受容体を分解するタンパク質)などのリポタンパク質代謝に関与する他の遺伝子に変化をもたらす可能性があります。
コレステロールの蓄積は有害である可能性があることが知られていますが、40歳より前に発生する心臓発作の最大XNUMX分のXNUMXは、診断されていない、または不適切に治療された家族の高コレステロール血症が原因であると推定されています。
より一般的なのは、素因となる遺伝的要因が存在する場合でも、環境要因(特に身体活動がない場合は脂肪が多い食事)によって引き起こされる多因子性高コレステロール血症です。
家族の高コレステロール血症が現れる形態
この病気は2つの異なる形態で現れる可能性があります:それほど深刻ではないもの(ヘテロ接合、1人ごとに500例)とより深刻なもの(ホモ接合、1人ごとに1,000,000例)1,2。
ヘテロ接合型は無症候性であることが多く、血中コレステロール値のみに基づいて診断されます。
LDL受容体の産生量が不十分なため、肝臓はLDLの除去に苦労しており、正常値と比較して血中濃度が2倍または3倍に増加します。
この形態は、成人期の心血管疾患のリスクを高める可能性があります。
ホモ接合型は、若い年齢でも心血管疾患の発症と、黄色腫(手の指関節とアキレス腱の黄色がかった結節)や黄色腫(まぶたとその周りの黄色がかったプレート)などの特徴的な脂肪の蓄積の存在によって特徴付けられます目)。
遺伝的欠陥は両親から受け継がれており、治療を受けていない場合の心臓発作のリスクは、15〜20歳前後ですでに観察されています。
実際、この状態では、肝臓は血液中に残っているリポタンパク質を代謝できず、それが蓄積して上記の機能障害を引き起こし、生命と両立しない状況を作り出します。
子供に病気が伝染する可能性は何ですか?
私たち一人一人が各遺伝子のXNUMXつのコピーを持っており、それぞれが親から継承されているため、XNUMXつの変更されたコピーとXNUMXつの正常なコピーを継承すると、ヘテロ接合型になります。
それどころか、両方の親から病気の遺伝子の両方のコピーを継承する場合、私たちはホモ接合型になります。 この形態は、両親が病気を引き起こす遺伝子を持っている場合にのみ発生し、したがって、それぞれが同じ遺伝子変化のコピーを送信できるため、非常にまれであると言わなければなりません。
家族性高コレステロール血症はヘテロ接合性病変と呼ばれ、XNUMXつの異なるタイプの遺伝子変化が各親からXNUMXつずつ遺伝するまれなケースで構成されます。
病気を発症するリスクのある子供は、早期に診断して治療する必要があります。 今日では、LDLR、ApoB、およびPCSK9遺伝子のエラー(遺伝子変化)を探す遺伝子検査を通じて病気を診断することが可能です。
したがって、家族の高コレステロール血症の形態のみが伝染します。
ただし、主に不適切なライフスタイル(食事によるコレステロールの蓄積)が原因で発生する多因子型は、遺伝的素因によっても特徴付けることができます。
実際、これらの被験者は、食事中の過剰な脂質を適切に補う身体の能力を損なう遺伝的欠陥を示している可能性があります。
たとえば、肝臓が食物と一緒に摂取されたコレステロールで飽和している場合、血液中を循環しているLDLを捕捉する受容体の生成が抑制される可能性があります。
状況は、それほど深刻ではありませんが、家族の高コレステロール血症の間に発生する状況と非常に似ています。
この場合、血中の総コレステロール濃度は通常より高く、通常は240〜350 mg / dlに設定されている通常の濃度と比較して、通常200〜240 mg / dlです。

好奇心:この病気は何世紀にもわたって受け継がれてきた可能性があります
モナリザの有名な絵を注意深く観察すると、モナリザはすでに若い年齢で家族の高コレステロール血症に苦しんでいた可能性があります。
実際、レオナルドはまた、手や目の近くの典型的な脂肪沈着物(現在は黄色腫および黄色腫として知られています)を忠実に描写しており、病理の存在を確実に示しています。
これが真実である場合、家族の高コレステロール血症は何世紀にもわたって保存されている病気であることが示されています(早くも1500年に記録された症例)。
今日この病気を治療する可能性は何ですか?
病気を治す可能性はその重症度に依存します。 また、危険因子(食事、喫煙、年齢、家族、高コレステロール血症の個人歴、他の疾患の存在)は、全体的な疾患の状況を悪化させる可能性があります。
家族の高コレステロール血症を治療するために、新世代の薬を使用することができますが、同時に、適切なライフスタイルの変更で行動する必要があります。
中等度の重症度(ヘテロ接合性家族高コレステロール血症)の場合、スタチンベースの薬物療法(LDLR受容体合成の増加を誘発するコレステロール合成阻害剤)およびエゼチミベ(コレステロール吸収阻害剤)またはPCSK9阻害剤(すなわち、その役割がLDLRを捕捉する肝臓受容体を破壊する)は、LDLR遺伝子の健康なコピーの活性を改善し、血中のコレステロールの蓄積を減らします。
最近承認されたのは、内因性コレステロール合成の過程に関与する分子であるATPクエン酸リアーゼ酵素を阻害することによって肝臓で作用する薬剤であるベンペド酸です。
この作用機序により、スタチンの作用部位の上流で作用するコレステロールの産生量に作用し、LDL受容体の発現を刺激して合成の低下を補うことができます。
スタチンとは異なり、ベンペド酸は骨格筋で活性がないため、スタチンに特有の望ましくないイベントが発生する可能性が低くなります。
この薬はまた、エゼチミブと関連して好ましい効果をもたらす可能性があります。
ホモ接合性家族性高コレステロール血症は、最近まで不治の病と見なされていました。
スタチンベースの治療は、この病状では効果的ではありません。 実際、内因性コレステロールの生成につながるメカニズムに作用するスタチンは、LDL受容体の合成を刺激することはできません。
これらの患者の体からLDLコレステロールを除去するために、血漿交換は、腎臓が機能していないときに透析で行われるのと同様に、脂肪を除去することによって血液をろ過することを可能にする技術です。
ただし、これは侵襲的な手順であり、患者の生活の質に悪影響を及ぼします。
家族の高コレステロール血症に関する最新の研究
近年、研究により、ホモ接合型の家族性高コレステロール血症にも対応する特定の薬剤が開発され、これに苦しむ患者の期待と生活の質が大幅に改善されています。
たとえば、ロミタピド(経口摂取)という薬は、これらの患者の血漿LDLコレステロールレベルを著しく低下させます。
ロミタピドはミクロソームトリグリセリド輸送タンパク質(MTP)を阻害し、これらの脂質をアポタンパク質B100と一緒に新生VLDLに取り込むことを可能にします。
その結果、アポタンパク質B100とLDLは、病気がLDL受容体の完全な欠如を引き起こした患者でも減少します。
別の新世代の薬剤は、LDLの形成に関与するB100アポタンパク質を分解することができるアンチセンスオリゴヌクレオチドであるミポメルセンであり、したがって後者の数を減らす。
エボロクマブとアリロクマブは、血中のPCSK9タンパク質の活性を阻害するXNUMXつのモノクローナル抗体です。
ただし、これらXNUMXつの薬が効果を発揮するには、LDL受容体の少なくとも一部が存在して機能している必要があります。
まだ臨床開発中の薬の中には、肝臓でのPCSK9タンパク質のDNA転写と合成をブロックする生物学的薬(いわゆるsiRNA)が含まれています。
最近、肝臓で合成されたタンパク質類似アンジオポエチン3(ANGPTL3)のモノクローナル抗体阻害剤であるエビナクマブで肯定的な結果が得られました。
この新薬は、LDL受容体が機能していない患者にも有効性を示しています3,4。
最近、患者のDNAに健康な遺伝子を導入する遺伝子治療が機能するかどうかを理解するためのいくつかの試みも行われています5。
家族性高コレステロール血症の可能な治療法のこのリストから、まれではありますが、この深刻な病気に苦しんでいるこれらの患者に解決策を提供するためにどれだけの研究が行われているかを理解することができ、致命的な結果のリスクを減らします。
しかし、新薬でいつも起こるように、それらの起こり得る副作用についての知識は経験(ファーマコビジランス)によってのみ明らかになるので、それらの慢性的な使用には注意を払うのが良いです、それで小児科でのそれらの使用は注意深く評価されるべきです、もちろん、これらの「小さな」患者に「通常の」生活を送る可能性を提供するという究極の目的を持っています。
家族の高コレステロール血症に関する記事の書誌的およびシトグラフィック参照
1年脂質異常症の管理に関するESC / EASガイドライン:心血管リスクを低減するための脂質修飾。 著者/タスクフォースメンバー; ESC実践ガイドライン委員会(CPG); ESC国立心臓学会。 アテローム性動脈硬化症。 2019年2019月; 290:140-205。 土井:10.1016 /j.atherosclerosis.2019.08.014。
2表現型から遺伝子型、管理までのまれな脂質異常症:欧州アテローム性動脈硬化症学会タスクフォースのコンセンサスステートメント。 Hegele RA、BorénJ、Ginsberg HN、Arca M、Averna M、Binder CJ、Calabresi L、Chapman MJ、Cuchel M、von Eckardstein A、Frikke-Schmidt R、Gaudet D、Hovingh GK、Kronenberg F、LütjohannD、Parhofer KG 、Raal FJ、Ray KK、Remaley AT、Stock JK、Stroes ES、TokgözoğluL、CatapanoAL。 ランセット糖尿病エンドクリノール。 2020年8月; 1(50):67-10.1016。 土井:2213 / S8587-19(30264)5-XNUMX。
3リポタンパク質(a)の低下-リポタンパク質アフェレーシスからアンチセンスオリゴヌクレオチドアプローチへ。 グレコMF、シルトリCR、コルシーニA、エジョフM、サンピエトロT、ルシカMJクリンメッド。 2020年3月9日; 7(2103):10.3390。 土井:9072103 / jcmXNUMX。
4LDL-コレステロール低下療法。 Pirillo A、Norata GD、Catapano AL.Handb ExpPharmacol。 2020年30月10.1007日。doi:164 / 2020_361_XNUMX。
5家族性高コレステロール血症に対する遺伝子ベースおよび細胞ベースの治療法のレビュー。 Hajighasemi S、Mahdavi Gorabi A、Bianconi V、Pirro M、Banach M、Ahmadi Tafti H、ReinerŽ、Sahebkar A.PharmacolRes。 2019年143月; 119:132-10.1016。 土井:2019.03.016 /j.phrs.XNUMX。