
Rodzinna hipercholesterolemia: co to jest i jak ją leczyć
Rodzinna hipercholesterolemia, rzadka choroba genetyczna, która powoduje poważne konsekwencje zdrowotne (np. Zawał serca i udar), wymagająca odpowiednich środków powstrzymujących, obejmujących odpowiednie leki i odpowiedniego stylu życia.
Co oznacza hipercholesterolemia? A co to znaczy „rodzina”?
Hipercholesterolemia to stan kliniczny charakteryzujący się nadmiernym poziomem cholesterolu we krwi.
Cholesterol jest składnikiem lipidów, zwykle przyjmowanych z dietą, ale może być również wytwarzany przez organizm.
Cholesterol jest bardzo ważny dla życia, ponieważ służy do tworzenia błon komórkowych (w celu zapewnienia ich funkcji), chroni neurony i nerwy czaszkowe.
Cholesterol jest również używany do syntezy innych cząsteczek, z których trzy to kwasy żółciowe (ważne dla trawienia), niektóre hormony i witamina D.
Tylko jeśli występuje powyżej tych wymagań, może spowodować poważne szkody.
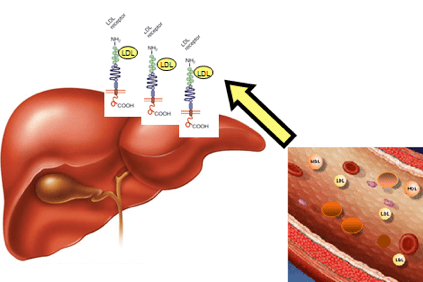
W przypadku hipercholesterolemii cholesterol gromadzi się we krwi w postaci lekkich lipoprotein (LDL), określonych agregatów tłuszczów i białek, zwanych także „złym cholesterolem”, które sprzyjają tworzeniu się blaszek w ścianie tętnic (blaszki miażdżycowe).
Dzieje się tak, gdy pacjenci mają tak wysoki poziom cholesterolu, że nie mogą go wyeliminować poprzez fizjologiczne mechanizmy wątroby.
Nagromadzenie LDL we krwi ostatecznie prowadzi do powstania miażdżycy (fizycznej przeszkody w prawidłowym przepływie krwi), co może prowadzić do poważniejszych konsekwencji, takich jak dusznica bolesna, zawał serca, udar, ale także w innych narządach, takich jak mózg, nerki, płuca i sama wątroba.
Hipercholesterolemia jest znana, gdy jest przenoszona na potomstwo
Wynika to z modyfikacji genu, który zawiera informacje potrzebne do wytworzenia białka wątroby, receptora LDLR (LDLR), który rozpoznaje LDL, usuwa go z krwiobiegu i transportuje do komórek wątroby, które następnie go usuwają. Ta zmiana genu powoduje gromadzenie się LDL we krwi.
Do chwili obecnej istnieje ponad 600 znanych zmian w genie LDL, które powodują rodzinną hipercholesterolemię.
Ponadto osoby z tym zaburzeniem mogą powodować zmiany w innych genach zaangażowanych w metabolizm lipoprotein, takich jak gen APOB (apoproteina B w LDL) i gen PCSK9 (białko, które degraduje receptory LDL).
Chociaż wiadomo, że akumulacja cholesterolu może być szkodliwa, szacuje się, że nawet jedna trzecia zawałów serca przed 40. rokiem życia jest spowodowana niezdiagnozowaną lub niewłaściwie leczoną rodzinną hipercholesterolemią.
Częściej występuje wieloczynnikowa hipercholesterolemia spowodowana czynnikami środowiskowymi (dieta bogata w tłuszcze, zwłaszcza jeśli jest związana z brakiem aktywności fizycznej), nawet w obecności predysponujących czynników genetycznych.
Formy, w których objawia się rodzinna hipercholesterolemia
Choroba ta może występować w 2 różnych formach: mniej poważnej (heterozygotyczna, 1 przypadek na 500 osób) i poważniejszej (homozygotyczna, 1 przypadek na 1,000,000 1,2 XNUMX osób) XNUMX.
Forma heterozygotyczna często przebiega bezobjawowo i jest diagnozowana tylko na podstawie poziomu cholesterolu we krwi.
Wątroba stara się wyeliminować LDL, ponieważ receptory LDL są wytwarzane w niewystarczającej liczbie, co powoduje 2 lub 3-krotny wzrost poziomu we krwi w porównaniu do wartości prawidłowych.
Ta forma może prowadzić do zwiększonego ryzyka chorób sercowo-naczyniowych w wieku dorosłym.
Postać homozygotyczna charakteryzuje się początkiem chorób układu krążenia już w młodym wieku i obecnością charakterystycznych nagromadzeń tłuszczu, takich jak ksantomy (żółtawe guzki na kostkach dłoni i ścięgnie Achillesa) i żółtawe płytki na powiekach i wokół oczy).
Wada genetyczna jest dziedziczona po obojgu rodzicach, a ryzyko zawału serca przy braku terapii obserwuje się już w wieku 15-20 lat.
W rzeczywistości w tym stanie wątroba nie metabolizuje lipoprotein, które pozostają we krwi, które następnie gromadzą się, prowadząc do opisanych powyżej dysfunkcji i stwarzając sytuację nie do pogodzenia z życiem.
Jakie są szanse przeniesienia choroby na dzieci?
Ponieważ każdy z nas ma dwie kopie każdego genu, każdy odziedziczony od rodzica, będziemy mieli postać heterozygotyczną, gdy odziedziczymy jedną zmienioną i jedną zdrową kopię.
Wręcz przeciwnie, będziemy mieli formę homozygotyczną, jeśli odziedziczymy obie kopie chorego genu od obojga rodziców. Trzeba powiedzieć, że ta forma jest bardzo rzadka, ponieważ występuje tylko wtedy, gdy oboje rodzice mają gen powodujący chorobę, a zatem każdy z nich może przekazać kopię tej samej zmiany genu.
Rodzinna hipercholesterolemia nazywana jest patologią heterozygotyczną występującą w rzadkich przypadkach, w których dziedziczone są dwa różne typy zmian genów, po jednym od każdego z rodziców.
Dzieci zagrożone rozwojem choroby należy wcześnie diagnozować i leczyć. Obecnie możliwe jest zdiagnozowanie choroby za pomocą testów genetycznych, które szukają błędów (zmian genów) w genach LDLR, ApoB i PCSK9.
Dlatego przenoszone są tylko postacie rodzinnej hipercholesterolemii.
Jednak formy wieloczynnikowe, które występują głównie z powodu nieprawidłowego trybu życia (kumulacji cholesterolu z dietą), mogą również charakteryzować się predyspozycjami genetycznymi.
W rzeczywistości u tych osobników mogą występować deficyty genetyczne, które upośledzają zdolność organizmu do odpowiedniego kompensowania nadmiaru lipidów w diecie.
Na przykład, gdy wątroba jest nasycona cholesterolem przyjmowanym z pożywieniem, produkcja receptorów wychwytujących LDL krążący we krwi może zostać zahamowana.
Sytuacja jest bardzo podobna do tej, która występuje podczas rodzinnej hipercholesterolemii, chociaż jest mniej dotkliwa.
W tym przypadku stężenie całkowitego cholesterolu we krwi jest powyżej normy i zwykle wynosi między 240 a 350 mg / dl w porównaniu do normalnego poziomu około 200 - 240 mg / dl.

Ciekawostka: ta choroba mogła być przenoszona na przestrzeni wieków
Uważna obserwacja słynnego obrazu Mona Lisy wskazuje na możliwość, że Mona Lisa już w młodym wieku cierpiała na rodzinną hipercholesterolemię.
W rzeczywistości Leonardo wiernie przedstawia również typowe złogi tłuszczu na dłoniach i w pobliżu oczu (obecnie znane jako ksantomy i ksantelazmy), które z pewnością wskazują na obecność patologii.
Jeśli to prawda, to wykazano, że rodzinna hipercholesterolemia jest chorobą utrwaloną na przestrzeni wieków (przypadek udokumentowany już w 1500 r.).
Jakie są dziś możliwości leczenia tej choroby?
Możliwość wyleczenia choroby zależy od jej ciężkości. Ponadto czynniki ryzyka (dieta, palenie tytoniu, wiek, rodzina i historia hipercholesterolemii, obecność innych chorób) mogą pogorszyć ogólny obraz choroby.
W leczeniu hipercholesterolemii rodzinnej można stosować leki nowej generacji, ale jednocześnie konieczne jest działanie z odpowiednią modyfikacją stylu życia.
W przypadkach o pośrednim nasileniu (heterozygotyczna rodzinna hipercholesterolemia), terapia lekowa oparta na statynach (inhibitory syntezy cholesterolu, które indukują zwiększoną syntezę receptora LDLR) oraz połączenie z ezetymibem (inhibitory wchłaniania cholesterolu) lub lekami hamującymi PCSK9 (tj. niszczą receptory wątrobowe wychwytujące LDLR) poprawia aktywność zdrowej kopii genu LDLR i zmniejsza gromadzenie się cholesterolu we krwi.
Niedawno zatwierdzony jest także kwas bempedonowy, lek działający w wątrobie poprzez hamowanie enzymu liazy cytrynianowej ATP, cząsteczki biorącej udział w procesie endogennej syntezy cholesterolu.
Ten mechanizm działania pozwala nam wpływać na ilość wytwarzanego cholesterolu, działając w górę od miejsca działania statyn i stymulować ekspresję receptorów LDL w celu kompensacji zmniejszonej syntezy.
W przeciwieństwie do statyn, kwas bempedonowy nie działa w mięśniach szkieletowych, co zmniejsza możliwość wystąpienia niepożądanych zdarzeń typowych dla statyn.
Lek ten może być również powiązany z ezetymibem z korzystnym działaniem.
Homozygotyczna rodzinna hipercholesterolemia była do niedawna uważana za chorobę nieuleczalną.
Terapia oparta na statynach nie jest skuteczna w tej patologii. W rzeczywistości statyny, które działają na mechanizmy prowadzące do produkcji endogennego cholesterolu, nie mogą stymulować syntezy receptorów LDL.
Aby wyeliminować cholesterol LDL z organizmu tych pacjentów, stosuje się plazmaferezę, technikę, która umożliwia filtrowanie krwi poprzez eliminację tłuszczów, podobnie jak w przypadku dializy, gdy nerki nie pracują.
Jest to jednak zabieg inwazyjny, który negatywnie wpływa na jakość życia pacjentów.
Najnowsze badania nad rodzinną hipercholesterolemią
W ostatnich latach badania doprowadziły do opracowania specyficznych leków również na homozygotyczną postać rodzinnej hipercholesterolemii, znacznie poprawiając oczekiwania i jakość życia chorych na nią.
Na przykład lek lomitapid (przyjmowany doustnie) prowadzi do znacznie niższych poziomów cholesterolu LDL w osoczu u tych pacjentów.
Lomitapid hamuje mikrosomalne białko transportujące trójglicerydy (MTP), które umożliwia włączenie tych lipidów wraz z apoproteiną B100 do powstających VLDL.
W rezultacie apoproteina B100 i LDL są obniżone nawet u pacjentów, u których choroba spowodowała całkowity brak receptora LDL.
Innym lekiem nowej generacji jest mipomersen, antysensowny oligonukleotyd zdolny do degradacji apoprotein B100, które biorą udział w tworzeniu LDL, zmniejszając w ten sposób liczbę tych ostatnich.
Ewolokumab i alirokumab to dwa przeciwciała monoklonalne, które hamują aktywność białka PCSK9 we krwi.
Jednak aby te dwa leki zadziałały, przynajmniej niewielka część receptorów LDL musi być obecna i funkcjonować.
Wśród leków, które wciąż są w fazie rozwoju klinicznego, jest włączenie leku biologicznego (tak zwanego siRNA), który blokuje transkrypcję DNA i syntezę białka PCSK9 w wątrobie.
Niedawno pozytywne wyniki uzyskano w przypadku evinakumabu, monoklonalnego przeciwciała inhibitora białka podobnego angiopoetyny 3 (ANGPTL3), cząsteczki syntetyzowanej przez wątrobę, której rolą jest zwiększenie poziomu cholesterolu-LLDL i trójglicerydów, zapobiegając ich degradacji.
Ten nowy lek okazał się również skuteczny u pacjentów, u których nie działa receptor LDL3,4.
Ostatnio podjęto również próby zrozumienia, czy terapia genowa, wraz z wprowadzeniem zdrowego genu do DNA pacjenta, może zadziałać5.
Z tej listy możliwych terapii rodzinnej hipercholesterolemii można zrozumieć, jak wiele badań prowadzi się w celu zapewnienia rozwiązań dla tych pacjentów, którzy, choć rzadko, są dotknięci tą poważną chorobą, zmniejszając ryzyko śmiertelnych konsekwencji.
Jednak jak to zawsze bywa w przypadku nowych leków, dobrze jest zachować ostrożność przy ich przewlekłym stosowaniu, gdyż wiedza o ich możliwych skutkach ubocznych będzie wyraźniejsza dopiero wraz z doświadczeniem (nadzór nad bezpieczeństwem farmakoterapii), dlatego ich zastosowanie w pediatrii powinno być dokładnie ocenione, oczywiście z ostatecznym celem zaoferowania tym „małym” pacjentom możliwości prowadzenia „normalnego” życia.
Źródła bibliograficzne i sitograficzne do artykułu o rodzinnej hipercholesterolemii
1 Wytyczne ESC / EAS 2019 dotyczące leczenia dyslipidemii: modyfikacja lipidów w celu zmniejszenia ryzyka sercowo-naczyniowego. Autorzy / członkowie grupy roboczej; Komitet ESC ds. Wytycznych praktycznych (CPG); Krajowe Towarzystwa Kardiologiczne ESC. Miażdżyca tętnic. Listopad 2019; 290: 140-205. doi: 10.1016 / j. miażdżyca.2019.08.014.
2 Rzadkie dyslipidemie, od fenotypu do genotypu i zarządzania: oświadczenie konsensusu grupy zadaniowej Europejskiego Towarzystwa Miażdżycy. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L, Catapano AL. Lancet Diabetes Endocrinol. 2020 styczeń; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoproteiny (a) Obniżenie poziomu aferezy lipoprotein do podejścia antysensownego oligonukleotydu. Greco MF, Sirtori CR, Corsini A, Ezhov M, Sampietro T, Ruscica MJ Clin Med. 2020 lipca 3; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 Terapia obniżająca poziom cholesterolu LDL. Pirillo A, Norata GD, Catapano AL. Handb Exp Pharmacol. 2020 Apr 30. doi: 10.1007 / 164_2020_361.
5 Przegląd terapii genowych i komórkowych w rodzinnej hipercholesterolemii. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, Pirro M, Banach M, Ahmadi Tafti H, Reiner Ž, Sahebkar A.Pharmacol Res. Maj 2019; 143: 119-132. doi: 10.1016 / j.phrs.2019.03.016.