
Породична хиперхолестеролемија: шта је то и како се лечити
Породична хиперхолестеролемија, ретка генетска болест која узрокује озбиљне здравствене последице (нпр. Срчани удар и мождани удар) које захтевају одговарајуће мере сузбијања уз одговарајуће лекове и одговарајући начин живота.
Шта значи хиперхолестеролемија? А шта значи породично?
Хиперхолестеролемија је клиничко стање које карактерише прекомерни холестерол у крви.
Холестерол је компонента липида, која се обично узима са исхраном, али коју тело такође може да произведе.
Холестерол је веома важан за живот јер се користи за формирање ћелијских мембрана (како би се осигурала њихова функција), штити неуроне и кранијалне живце.
Холестерол се користи и за синтезу других молекула, од којих су три жучне киселине (важне за варење), неки хормони и витамин Д.
Само ако је присутан изнад ових захтева, може проузроковати озбиљну штету.
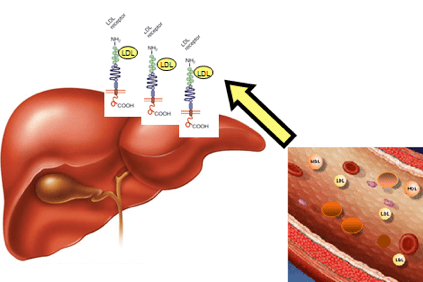
У случају хиперхолестеролемије, холестерол се акумулира у крви у облику лаких липопротеина (ЛДЛ), одређених агрегата масти и протеина, такође названих „лош холестерол“ који поспешују стварање плакова у зиду артерије (атеросклеротични плакови).
То се дешава када пацијенти имају тако висок ниво холестерола да га не могу елиминисати путем физиолошких механизама јетре.
Акумулација ЛДЛ у крви на крају доводи до стварања атерома (физичка препрека нормалном протоку крви) што може довести до озбиљнијих последица попут ангине, срчаног удара, можданог удара, али и у другим органима као што је мозак, бубрега, плућа и саме јетре.
Хиперхолестеролемија је позната када се преноси на потомство
То је због модификација гена који садржи информације да би се створио протеин јетре, ЛДЛР (ЛДЛР) рецептор, који препознаје ЛДЛ, уклања га из крвотока и транспортује до ћелија јетре, које га затим уклањају. Ова промена гена узрокује акумулацију ЛДЛ у крви.
До данас је познато више од 600 промена у ЛДЛ гену које узрокују породичну хиперхолестеролемију.
Такође, људи са овим поремећајем могу донети промене у другим генима који су укључени у метаболизме липопротеина, попут АПОБ гена (апопротеин Б у ЛДЛ) и гена ПЦСК9 (протеин који разграђује ЛДЛ рецепторе).
Иако је познато да акумулација холестерола може бити штетна, процењује се да је до једне трећине срчаних удара који су се јавили пре 40. године живота услед недијагностиковане или неадекватно лечене породичне хиперхолестеролемије.
Чешћа је мултифакторска хиперхолестеролемија узрокована факторима околине (дијета богата масноћама, посебно ако је повезана са физичком неактивношћу), чак и у присуству генетских фактора који предиспонирају.
Облици у којима се манифестује породична хиперхолестеролемија
Ова болест се може представити у 2 различита облика: мање озбиљном (хетерозиготном, 1 случај на 500 јединки) и озбиљнијим (хомозиготним, 1 случај на 1,000,000 јединки) 1,2.
Хетерозиготни облик је често асимптоматски и дијагностикује се само на основу нивоа холестерола у крви.
Јетра се бори да елиминише ЛДЛ, јер се ЛДЛ рецептори производе у недовољном броју, што резултира двоструким или троструким повећањем нивоа крви у поређењу са нормалним вредностима.
Овај облик може довести до повећаног ризика од кардиоваскуларних болести у одраслој доби.
Хомозиготни облик карактерише почетак кардиоваскуларних болести чак и у младим годинама и присуство карактеристичних накупина масти као што су ксантоми (жућкасти чворићи на зглобовима руку и Ахилова тетива) и ксантелазма (жућкасте плочице на капцима и око очи).
Генетски дефект наслеђује се од оба родитеља, а ризик од срчаног удара у одсуству терапије већ се примећује око 15-20 година.
Заправо, у овом стању јетра не успева да метаболише липопротеине који остају у крви, који се затим акумулирају, што доводи до горе описаних дисфункција и ствара ситуацију некомпатибилну са животом.
Какве су шансе за пренос болести на децу?
Будући да свако од нас има две копије сваког гена, свака наслеђена од родитеља, имаћемо хетерозиготни облик када наследимо једну измењену и једну здраву копију.
Супротно томе, имаћемо хомозиготни облик ако обе копије наследимо од обоје родитеља. Мора се рећи да је овај облик врло редак, јер се јавља само када оба родитеља имају ген који узрокује болест и зато сваки од њих може да пренесе копију исте промене гена.
Породична хиперхолестеролемија назива се хетерозиготна патологија састављена у ретким случајевима у којима се наслеђују две различите врсте промена гена, по једна од сваког родитеља.
Деца у ризику од развоја болести морају се рано дијагностиковати и лечити. Данас је могуће дијагнозирати болест генетским тестовима који траже грешке (промене гена) на генима ЛДЛР, АпоБ и ПЦСК9.
Стога су преносиви само облици породичне хиперхолестеролемије.
Међутим, мултифакторски облици који се јављају углавном због неправилног начина живота (акумулација холестерола у исхрани), такође могу бити окарактерисани генетском предиспозицијом.
У ствари, код ових субјеката могу бити присутни генетски дефицити који угрожавају способност тела да адекватно надокнади вишак липида у исхрани.
На пример, када је јетра засићена холестеролом узетим са храном, производња рецептора који хватају ЛДЛ који циркулише у крви може бити сузбијена.
Ситуација је врло слична ситуацији која се јавља током породичне хиперхолестеролемије, иако мање тешка.
У овом случају, концентрација укупног холестерола у крви је изнад нормалне и обично је између 240 и 350 мг / дл у поређењу са нормалном која је уобичајено постављена око 200 - 240 мг / дл.

Куриозитет: ова болест се можда преносила вековима
Пажљиво посматрање чувене слике Мона Лизе показује могућност да је Мона Лиза већ у младости патила од породичне хиперхолестеролемије.
У ствари, Леонардо такође верно приказује типичне наслаге масти на рукама и у близини очију (данас познате као ксантоми и ксантелазма) које са сигурношћу указују на присуство патологије.
Ако је ово тачно, показује се да је породична хиперхолестеролемија болест која се очувала вековима (случај документован већ 1500).
Какве су могућности за лечење ове болести данас?
Могућност излечења болести зависи од њене тежине. Такође, фактори ризика (дијета, пушење, старост, породица и лична историја хиперхолестеролемије, присуство других болести) могу погоршати укупну слику болести.
За лечење породичне хиперхолестеролемије могу се користити лекови нове генерације, али истовремено је потребно деловати са одговарајућом модификацијом начина живота.
У случајевима средње тежине (хетерозиготна породична хиперхолестеролемија), терапија лековима заснована на статинима (инхибитори синтезе холестерола који индукују повећану синтезу ЛДЛР рецептора) и комбинација са езетимибом (инхибитори апсорпције холестерола) или ПЦСК9 (тј. Инхибитори протеина ПЦСК9 чија је улога да уништи рецепторе јетре који хватају ЛДЛР) побољшава активност здраве копије гена ЛДЛР и смањује акумулацију холестерола у крви.
Недавно је одобрена и бемпедојска киселина, лек који делује у јетри инхибирајући ензим АТП цитрат лиазу, молекул укључен у процес ендогене синтезе холестерола.
Овај механизам деловања омогућава нам да делујемо на количину произведеног холестерола, делујући узводно од места деловања статина, и да стимулишемо експресију ЛДЛ рецептора да надокнадимо смањену синтезу.
За разлику од статина, бемпедојска киселина није активна у скелетним мишићима, што смањује могућност појаве нежељених догађаја типичних за статине.
Овај лек се такође може повезати са езетимибом са повољним ефектима.
Хомозиготна породична хиперхолестеролемија се донедавно сматрала неизлечивом болешћу.
Терапија заснована на статинима није ефикасна у овој патологији. У ствари, статини који делују на механизме који доводе до стварања ендогеног холестерола не могу стимулисати синтезу ЛДЛ рецептора.
Да би се елиминисао ЛДЛ холестерол из тела ових пацијената, плазмафереза, техника која омогућава филтрирање крви уклањањем масти, слично ономе што се ради са дијализом када бубрези не раде.
Међутим, ово је инвазивни поступак са негативним утицајем на квалитет живота пацијената.
Најновије истраживање породичне хиперхолестеролемије
Последњих година истраживања су довела до развоја специфичних лекова такође за хомозиготни облик породичне хиперхолестеролемије, у великој мери побољшавајући очекивања и квалитет живота пацијената који пате од ње.
На пример, лек ломитапид (узет орално) доводи до знатно нижих нивоа ЛДЛ-холестерола у плазми код ових пацијената.
Ломитапид инхибира микросомски транспортни протеин триглицерида (МТП) који омогућава уградњу ових липида заједно са апопротеином Б100 у новонастале ВЛДЛ.
Као резултат, апопротеин Б100 и ЛДЛ су смањени чак и код пацијената код којих је болест проузроковала потпуно одсуство ЛДЛ рецептора.
Још један лек нове генерације је мипомерсен, антисенсе олигонуклеотид способан да разгради Б100 апопротеине који учествују у стварању ЛДЛ-а, смањујући тако њихов број.
Еволокумаб и алирокумаб су два моноклонска антитела која инхибирају активност протеина ПЦСК9 у крви.
Међутим, да би ова два лека ступила на снагу, бар мали део ЛДЛ рецептора мора бити присутан и функционисати.
Међу лековима који су још увек у клиничком развоју је укључивање биолошког лека (тзв. СиРНА) који блокира транскрипцију ДНК и синтезу протеина ПЦСК9 у јетри.
Позитивни резултати недавно су постигнути са евинакумабом, инхибитором моноклонских антитела протеина сличног ангиопоиетину 3 (АНГПТЛ3), молекулом који синтетише јетра чија је улога повећање нивоа холестерола-ЛЛДЛ и триглицерида спречавајући њихову разградњу.
Овај нови лек такође је показао ефикасност код пацијената код којих ЛДЛ рецептор не функционише3,4.
Недавно су учињени и неки покушаји да се разуме да ли би генска терапија, увођењем здравог гена у ДНК пацијента, могла да функционише5.
Из ове листе могућих терапија породичне хиперхолестеролемије могуће је разумети колико се истраживања спроводи како би се обезбедила решења за ове пацијенте који су, иако ретки, погођени овом озбиљном болешћу, смањујући ризик од фаталних последица.
Међутим, као и увек са новим лековима, добро је упозорити на њихову хроничну употребу, јер ће знање о њиховим могућим нежељеним ефектима бити јасније само са искуством (фармаковигиланца), па њихову употребу у педијатрији треба пажљиво проценити, наравно са крајњим циљем да се тим „малим“ пацијентима пружи могућност да воде „нормалан“ живот.
Библиографске и ситографске референце за чланак о породичној хиперхолестеролемији
1 2019 ЕСЦ / ЕАС смернице за лечење дислипидемија: Модификација липида ради смањења кардиоваскуларног ризика. Аутори / чланови радне групе; ЕСЦ комитет за смернице за праксу (ЦПГ); ЕСЦ Национална кардиолошка друштва. Атеросклероза. 2019. новембар; 290: 140-205. дои: 10.1016 / ј.атеросклероза.2019.08.014.
2 Ретке дислипидемије, од фенотипа до генотипа до управљања: изјава консензуса радне групе Европског удружења за атеросклерозу. Хегеле РА, Борен Ј, Гинсберг ХН, Арца М, Аверна М, Биндер ЦЈ, Цалабреси Л, Цхапман МЈ, Цуцхел М, вон Ецкардстеин А, Фрикке-Сцхмидт Р, Гаудет Д, Ховингх ГК, Кроненберг Ф, Лутјоханн Д, Пархофер КГ , Раал ФЈ, Раи КК, Ремалеи АТ, Стоцк ЈК, Строес ЕС, Токгозоглу Л, Цатапано АЛ. Ланцет Диабетес Ендоцринол. 2020. јануар; 8 (1): 50-67. дои: 10.1016 / С2213-8587 (19) 30264-5.
3 Липопротеин (а) Спуштање - од аферезе липопротеина до антисенсе олигонуклеотидног приступа. Грецо МФ, Сиртори ЦР, Цорсини А, Езхов М, Сампиетро Т, Русцица МЈ Цлин Мед. 2020. 3. јул; 9 (7): 2103. дои: 10.3390 / јцм9072103.
4 Терапија за смањење ЛДЛ-холестерола. Пирилло А, Нората ГД, Цатапано АЛ.Хандб Екп Пхармацол. 2020. април 30. дои: 10.1007 / 164_2020_361.
5 Преглед генетске и ћелијске терапије породичне хиперхолестеролемије. Хајигхасеми С, Махдави Гораби А, Бианцони В, Пирро М, Банацх М, Ахмади Тафти Х, Реинер Ж, Сахебкар А.Пхармацол Рес. 2019. мај; 143: 119-132. дои: 10.1016 / ј.пхрс.2019.03.016.