
Familienhypercholesterinämie: Was es ist und wie man es behandelt
Familiäre Hypercholesterinämie, eine seltene genetisch bedingte Krankheit, die schwerwiegende gesundheitliche Folgen hat (z. B. Herzinfarkt und Schlaganfall) und geeignete Eindämmungsmaßnahmen mit geeigneten Medikamenten und einem angemessenen Lebensstil erfordert.
Was bedeutet Hypercholesterinämie? Und was bedeutet familiär?
Hypercholesterinämie ist eine klinische Erkrankung, die durch übermäßiges Cholesterin im Blut gekennzeichnet ist.
Cholesterin ist ein Bestandteil von Lipiden, die normalerweise über die Nahrung aufgenommen werden, aber auch vom Körper produziert werden können.
Cholesterin ist sehr wichtig für das Leben, da es zur Bildung von Zellmembranen (um deren Funktion sicherzustellen) verwendet wird und Neuronen und Hirnnerven schützt.
Cholesterin wird auch zur Synthese anderer Moleküle verwendet, von denen drei Gallensäuren (wichtig für die Verdauung), einige Hormone und Vitamin D sind.
Nur wenn diese Anforderungen erfüllt sind, kann dies zu ernsthaften Schäden führen.
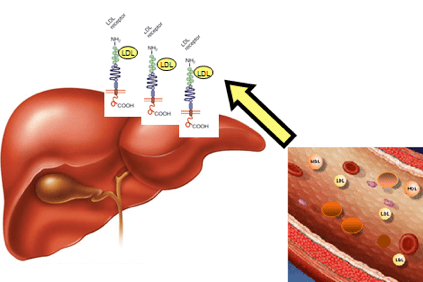
Bei Hypercholesterinämie reichert sich Cholesterin im Blut in Form von leichten Lipoproteinen (LDL) an, bestimmten Aggregaten von Fetten und Proteinen, auch „schlechtes Cholesterin“ genannt, die die Bildung von Plaques in der Arterienwand fördern (atherosklerotische Plaques).
Dies geschieht, wenn Patienten einen so hohen Cholesterinspiegel haben, dass sie ihn nicht durch die physiologischen Mechanismen der Leber eliminieren können.
Die Anreicherung von LDL im Blut führt schließlich zur Bildung des Atheroms (eine physische Behinderung des normalen Blutflusses), was zu schwerwiegenderen Folgen wie Angina, Herzinfarkt, Schlaganfall, aber auch in anderen Organen wie dem Gehirn führen kann. Nieren, Lunge und die Leber selbst.
Hypercholesterinämie ist bekannt, wenn sie auf Nachkommen übertragen wird
Dies ist auf Modifikationen eines Gens zurückzuführen, das die Informationen zur Herstellung eines Leberproteins enthält. Der LDLR (LDLR) -Rezeptor, der LDL erkennt, entfernt es aus dem Blutkreislauf und transportiert es zu den Leberzellen, die es dann entfernen. Diese Genveränderung verursacht eine LDL-Akkumulation im Blut.
Bis heute sind mehr als 600 Veränderungen im LDL-Gen bekannt, die eine familiäre Hypercholesterinämie verursachen.
Menschen mit dieser Störung können auch Veränderungen in anderen Genen hervorrufen, die am Lipoproteinstoffwechsel beteiligt sind, wie dem APOB-Gen (Apoprotein B in LDL) und dem PCSK9-Gen (einem Protein, das LDL-Rezeptoren abbaut).
Obwohl bekannt ist, dass die Cholesterinakkumulation schädlich sein kann, wird geschätzt, dass bis zu einem Drittel der vor dem 40. Lebensjahr auftretenden Herzinfarkte auf eine nicht diagnostizierte oder unangemessen behandelte familiäre Hypercholesterinämie zurückzuführen sind.
Häufiger ist eine multifaktorielle Hypercholesterinämie, die durch Umweltfaktoren (eine fettreiche Ernährung, insbesondere wenn sie mit körperlicher Inaktivität verbunden ist) verursacht wird, selbst wenn prädisponierende genetische Faktoren vorliegen.
Die Formen der familiären Hypercholesterinämie manifestieren sich
Diese Krankheit kann in zwei verschiedenen Formen auftreten: eine weniger schwerwiegende (heterozygot, 2 Fall alle 1 Personen) und eine schwerwiegendere (homozygot, 500 Fall alle 1 Personen) 1,000,000.
Die heterozygote Form ist oft asymptomatisch und wird nur anhand des Cholesterinspiegels im Blut diagnostiziert.
Die Leber kämpft darum, LDL zu eliminieren, da LDL-Rezeptoren in unzureichender Anzahl produziert werden, was zu einem 2- oder 3-fachen Anstieg der Blutspiegel im Vergleich zu normalen Werten führt.
Diese Form kann im Erwachsenenalter zu einem erhöhten Risiko für Herz-Kreislauf-Erkrankungen führen.
Die homozygote Form ist gekennzeichnet durch das Auftreten von Herz-Kreislauf-Erkrankungen bereits in jungen Jahren und das Vorhandensein charakteristischer Fettansammlungen wie Xanthome (gelbliche Knötchen an den Fingerknöcheln und der Achillessehne) und Xanthelasma (gelbliche Platten an den Augenlidern und in der Umgebung) die Augen).
Der genetische Defekt wird von beiden Elternteilen vererbt und das Risiko eines Herzinfarkts ohne Therapie wird bereits im Alter von 15 bis 20 Jahren beobachtet.
Tatsächlich kann die Leber in diesem Zustand die im Blut verbleibenden Lipoproteine nicht metabolisieren, die sich dann ansammeln, was zu den oben beschriebenen Funktionsstörungen führt und eine mit dem Leben unvereinbare Situation schafft.
Wie hoch sind die Chancen, dass die Krankheit auf Kinder übertragen wird?
Da jeder von uns zwei Kopien jedes Gens hat, die jeweils von einem Elternteil geerbt wurden, haben wir die heterozygote Form, wenn wir eine veränderte und eine gesunde Kopie erben.
Im Gegenteil, wir werden die homozygote Form haben, wenn wir beide Kopien des erkrankten Gens von beiden Elternteilen erben. Es muss gesagt werden, dass diese Form sehr selten ist, da sie nur auftritt, wenn beide Elternteile das Gen haben, das die Krankheit verursacht, und daher jeder von ihnen eine Kopie derselben Genveränderung übertragen kann.
Familiäre Hypercholesterinämie wird als heterozygote Pathologie bezeichnet, die in den seltenen Fällen auftritt, in denen zwei verschiedene Arten von Genveränderungen vererbt werden, eine von jedem Elternteil.
Kinder, bei denen das Risiko besteht, an der Krankheit zu erkranken, müssen frühzeitig diagnostiziert und behandelt werden. Heute ist es möglich, die Krankheit durch Gentests zu diagnostizieren, bei denen nach Fehlern (Genveränderungen) bei LDLR-, ApoB- und PCSK9-Genen gesucht wird.
Daher sind nur die Formen der familiären Hypercholesterinämie übertragbar.
Multifaktorielle Formen, die hauptsächlich aufgrund eines falschen Lebensstils (Anreicherung von Cholesterin mit der Nahrung) auftreten, können jedoch auch durch eine genetische Veranlagung gekennzeichnet sein.
Tatsächlich können diese Probanden genetische Defizite aufweisen, die die Fähigkeit des Körpers beeinträchtigen, überschüssiges Lipid in der Ernährung angemessen zu kompensieren.
Wenn zum Beispiel die Leber mit Cholesterin gesättigt ist, das mit der Nahrung eingenommen wird, kann die Produktion von Rezeptoren, die im Blut zirkulierendes LDL einfangen, unterdrückt werden.
Eine Situation ist der bei familiärer Hypercholesterinämie sehr ähnlich, wenn auch weniger schwerwiegend.
In diesem Fall liegt die Konzentration des Gesamtcholesterins im Blut über dem Normalwert und liegt normalerweise zwischen 240 und 350 mg / dl im Vergleich zu der normalen Konzentration, die üblicherweise auf 200 bis 240 mg / dl eingestellt ist.

Eine Kuriosität: Diese Krankheit wurde möglicherweise im Laufe der Jahrhunderte weitergegeben
Eine sorgfältige Beobachtung des berühmten Gemäldes der Mona Lisa zeigt die Möglichkeit, dass Mona Lisa bereits in jungen Jahren an familiärer Hypercholesterinämie litt.
Tatsächlich zeigt Leonardo auch die typischen Fettablagerungen an den Händen und in der Nähe der Augen (heute als Xanthome und Xanthelasma bekannt), die das Vorhandensein der Pathologie mit Sicherheit anzeigen.
Wenn dies zutrifft, wird gezeigt, dass familiäre Hypercholesterinämie eine Krankheit ist, die über Jahrhunderte erhalten geblieben ist (ein Fall, der bereits 1500 dokumentiert wurde).
Welche Möglichkeiten gibt es heute, um diese Krankheit zu behandeln?
Die Möglichkeit, die Krankheit zu heilen, hängt von ihrer Schwere ab. Auch Risikofaktoren (Ernährung, Rauchen, Alter, Familie und persönliche Vorgeschichte von Hypercholesterinämie, Vorhandensein anderer Krankheiten) können das Gesamtbild der Krankheit verschlechtern.
Zur Behandlung von familiärer Hypercholesterinämie können Medikamente der neuen Generation verwendet werden, gleichzeitig ist es jedoch erforderlich, mit einer angemessenen Änderung des Lebensstils zu handeln.
In Fällen mittlerer Schwere (heterozygote Familienhypercholesterinämie), statinbasierte Arzneimitteltherapie (Cholesterinsynthesehemmer, die eine erhöhte LDLR-Rezeptorsynthese induzieren) und die Kombination mit Ezetimib (Cholesterinabsorptionshemmer) oder PCSK9-Hemmstoffen (dh PCSK9-Proteininhibitoren, deren Rolle es ist, Leberrezeptoren zerstören, die LDLR einfangen) verbessert die Aktivität der gesunden Kopie des LDLR-Gens und verringert die Akkumulation von Cholesterin im Blut.
Kürzlich zugelassen wurde auch Bempedosäure, ein Medikament, das in der Leber wirkt, indem es das Enzym ATP-Citrat-Lyase hemmt, ein Molekül, das am Prozess der endogenen Cholesterinsynthese beteiligt ist.
Dieser Wirkungsmechanismus ermöglicht es uns, auf die Menge des produzierten Cholesterins einzuwirken, die stromaufwärts vom Wirkort der Statine wirkt, und die Expression von LDL-Rezeptoren zu stimulieren, um die reduzierte Synthese zu kompensieren.
Im Gegensatz zu Statinen ist Bempedosäure im Skelettmuskel nicht aktiv, was die Möglichkeit von unerwünschten Ereignissen verringert, die für Statine typisch sind.
Dieses Medikament kann auch mit Ezetimib mit günstigen Wirkungen assoziiert sein.
Homozygote familiäre Hypercholesterinämie wurde bis vor kurzem als unheilbare Krankheit angesehen.
Eine auf Statin basierende Therapie ist bei dieser Pathologie nicht wirksam. Tatsächlich können Statine, die auf die Mechanismen einwirken, die zur Produktion von endogenem Cholesterin führen, die Synthese von LDL-Rezeptoren nicht stimulieren.
Um LDL-Cholesterin aus dem Körper dieser Patienten zu eliminieren, Plasmapherese, eine Technik, die es ermöglicht, das Blut durch Eliminierung von Fetten zu filtern, ähnlich wie bei der Dialyse, wenn die Nieren nicht funktionieren.
Dies ist jedoch ein invasives Verfahren, das sich negativ auf die Lebensqualität der Patienten auswirkt.
Die neuesten Forschungsergebnisse zur familiären Hypercholesterinämie
In den letzten Jahren hat die Forschung zur Entwicklung spezifischer Medikamente auch für die homozygote Form der familiären Hypercholesterinämie geführt, was die Erwartung und Lebensqualität von Patienten, die darunter leiden, erheblich verbessert.
Beispielsweise führt das Medikament Lomitapid (oral eingenommen) bei diesen Patienten zu deutlich niedrigeren Plasma-LDL-Cholesterinspiegeln.
Lomitapid hemmt ein mikrosomales Triglyceridtransportprotein (MTP), das den Einbau dieser Lipide zusammen mit dem Apoprotein B100 in entstehende VLDLs ermöglicht.
Infolgedessen sind das Apoprotein B100 und LDL selbst bei Patienten reduziert, bei denen die Krankheit das völlige Fehlen des LDL-Rezeptors verursacht hat.
Ein weiteres Medikament der neuen Generation ist Mipomersen, ein Antisense-Oligonukleotid, das die an der Bildung von LDL beteiligten B100-Apoproteine abbauen und so deren Anzahl verringern kann.
Evolocumab und Alirocumab sind zwei monoklonale Antikörper, die die Aktivität des PCSK9-Proteins im Blut hemmen.
Damit diese beiden Medikamente wirken, muss jedoch mindestens ein kleiner Teil der LDL-Rezeptoren vorhanden sein und funktionieren.
Zu den noch in der klinischen Entwicklung befindlichen Arzneimitteln gehört der Einschluss eines biologischen Arzneimittels (sogenannte siRNA), das die DNA-Transkription und -Synthese des PCSK9-Proteins in der Leber blockiert.
Positive Ergebnisse wurden kürzlich mit Evinacumab erzielt, einem monoklonalen Antikörperinhibitor des Proteins ähnlich Angiopoietin 3 (ANGPTL3), einem von der Leber synthetisierten Molekül, dessen Aufgabe es ist, die Cholesterin-LLDL- und Triglyceridspiegel zu erhöhen, um deren Abbau zu verhindern.
Dieses neue Medikament hat auch Wirksamkeit bei Patienten gezeigt, bei denen der LDL-Rezeptor nicht funktioniert3,4.
In letzter Zeit wurden auch einige Versuche unternommen, um zu verstehen, ob eine Gentherapie mit der Einführung des gesunden Gens in die DNA des Patienten funktionieren könnte5.
Aus dieser Liste möglicher Therapien für familiäre Hypercholesterinämie ist ersichtlich, wie viel Forschung betrieben wird, um Lösungen für diese Patienten bereitzustellen, die zwar selten, aber von dieser schweren Krankheit betroffen sind, wodurch das Risiko tödlicher Folgen verringert wird.
Wie immer bei neuen Arzneimitteln ist es jedoch ratsam, bei ihrem chronischen Gebrauch ein Wort der Vorsicht walten zu lassen, da das Wissen über ihre möglichen Nebenwirkungen nur mit der Erfahrung (Pharmakovigilanz) klarer wird. Daher sollte ihr Einsatz in der Pädiatrie sorgfältig abgewogen werden. Natürlich mit dem ultimativen Ziel, diesen „kleinen“ Patienten die Möglichkeit zu bieten, ein „normales“ Leben zu führen.
Bibliografische und sitografische Verweise auf den Artikel zur familiären Hypercholesterinämie
1 2019 ESC / EAS-Richtlinien zur Behandlung von Dyslipidämien: Lipidmodifikation zur Reduzierung des kardiovaskulären Risikos. Autoren / Task Force-Mitglieder; ESC-Ausschuss für Leitlinien für die Praxis (CPG); ESC National Cardiac Societies. Atherosklerose. 2019 Nov; 290: 140–205. doi: 10.1016 / j.atherosclerosis.2019.08.014.
2 Seltene Dyslipidämien, vom Phänotyp über den Genotyp bis zum Management: eine Konsenserklärung der Task Force der European Atherosclerosis Society. Hegele RA, Borén J, Ginsberg HN, Arca M, Averna M, Binder CJ, Calabresi L, Chapman MJ, Cuchel M, von Eckardstein A, Frikke-Schmidt R, Gaudet D, Hovingh GK, Kronenberg F, Lütjohann D, Parhofer KG , Raal FJ, Ray KK, Remaley AT, Stock JK, Stroes ES, Tokgözoğlu L., Catapano AL. Lancet Diabetes Endocrinol. 2020 Jan; 8 (1): 50-67. doi: 10.1016 / S2213-8587 (19) 30264-5.
3 Lipoprotein (a) Absenkung von der Lipoproteinapherese zum Antisense-Oligonukleotid-Ansatz. Greco MF, Sirtori CR, Corsini A., Ezhov M., Sampietro T., Ruscica MJ Clin Med. 2020, 3. Juli; 9 (7): 2103. doi: 10.3390 / jcm9072103.
4 LDL-cholesterinsenkende Therapie. Pirillo A, Norata GD, Catapano AL. Handb Exp Pharmacol. 2020 30. April doi: 10.1007 / 164_2020_361.
5 Ein Überblick über gen- und zellbasierte Therapien bei familiärer Hypercholesterinämie. Hajighasemi S., Mahdavi Gorabi A., Bianconi V., Pirro M., Banach M., Ahmadi Tafti H., Reiner ®, Sahebkar A. Pharmacol Res. 2019 May; 143: 119 & ndash; 132. doi: 10.1016 / j.phrs.2019.03.016.
Lesen Sie auch:
Lesen Sie den italienischen Artikel