
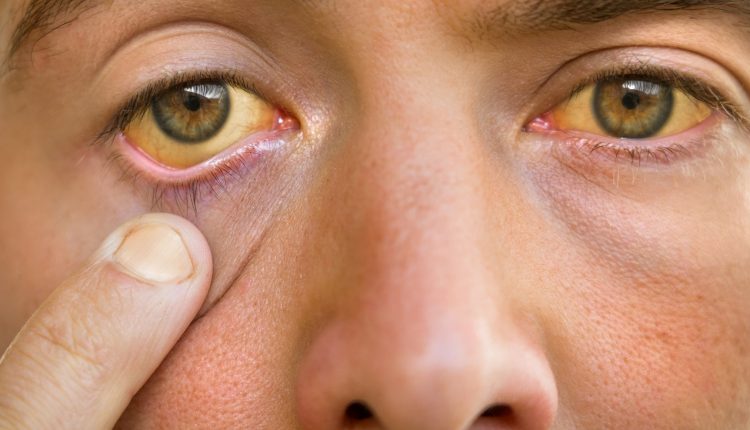
Hva er Gilberts syndrom
Gilberts syndrom er en familiær og arvelig patofysiologisk tilstand, med liten klinisk relevans og med godartet prognose, karakterisert ved gulsott eller leversubarterie, forårsaket av en medfødt endring av bilirubinmetabolismen og påfølgende økning i bilirubinemi i blodet, dvs. hyperbilirubinemi (med skleral gulsott eller subarterie)
Hyperbilirubinemi manifesterer seg med gulsott og er differensiert etter ulike typer.
Gulsott er klassifisert i tre arter: pre-hepatisk, hepatisk og post-hepatisk.
Gilberts syndrom er en gulsott (eller sub-aterektomi hvis mindre manifest) av levertypen, med varierende intensitet
Det avhenger av en endret funksjon i hepatocytten, leverens hovedcelle; det er en medfødt og familiær enzymatisk defekt, som fører til en dysfunksjon og reduksjon av den normale prosessen med glukurono-konjugering av bilirubin, noe som resulterer i et overskudd av indirekte bilirubin i serumet og i sammensetningen av galle.
Bilirubin er sluttproduktet av normal hemnedbrytning, det kommer fra katabolismen av hem som finnes i de aldrende røde blodcellene (hver rød blodcelle varer ca. 100 dager, i Gilberts enda mindre); bilirubinproduksjonen foregår i retikuloendotelsystemet, hovedsakelig milt og benmarg, hvoretter det tas opp av hepatocytter i leveren som utsetter den for glukurono-konjugasjonsprosessen, som er defekt i Gilberts syndrom.
Dermed frigjøres bilirunin i en indirekte eller "ukonjugert" form.
Denne forstyrrelsen av bilirubinmetabolismen kan ha varierende alvorlighetsgrad, fra mild som ved Gilbert syndrom, til alvorlig som ved forhøyet hyperbilirubinemi av Crigler-Najjar I og II syndromer, heldigvis mindre hyppig og sjelden.
Hvordan Gilberts syndrom manifesterer seg
Gilberts syndrom er en ganske hyppig tilstand (omtrent 5 – 8 % av den italienske befolkningen).
Lider har en vanligvis fluktuerende økning i serumbilirubinemi, spesielt indirekte eller ukonjugert bilirubinemi (noen ganger også direkte bilirubinemi, men i dette tilfellet ikke utbredt).
Hyppigheten av bilirubinemi og ukonjugert bilirubin er generelt svært variabel, og er ofte innenfor normalområdet hos mange individer med Gilbert syndrom.
Det er en arvelig endring som ikke skaper noen form for problemer med hverken kvaliteten eller lengden på livet til pasienter.
Under visse forhold (feber, infeksjoner, interkurver, stress, alkoholmisbruk, fysisk anstrengelse, faste) kan det oppstå en markant økning, selv over verdien på 2.5 – 5 mg/100 ml bilirubinemi, som imidlertid umiddelbart går tilbake til normalen. når disse spesielle, vanligvis midlertidige, situasjonene opphører.
Diagnose av Gilberts syndrom
Gilbert syndrom oppdages i forsøkspersonen for det meste ved en tilfeldighet, mest under laboratorieundersøkelser utført for rutinemessige eller sporadiske kontroller.
Alderen for debut av en første gulsott eller subiktal episode er vanligvis rundt 15-18 år.
Hos 40 % av personer med Gilbert syndrom er det en liten reduksjon i levetiden for røde blodlegemer, noe som kan føre til mistanke om hemolyse.
Diagnosen Gilberts syndrom krever først utelukkelse av enhver annen hepatocellulær, biliær, hemolytisk sykdom som kan forårsake (eller fremkalle) gulsott; i tillegg en økning i bilirubinemi, hovedsakelig indirekte bilirubinemi minst tre ganger etter hverandre, to eller tre funn av økte serumverdier med noen måneders mellomrom, samtidig med absolutt normalitet av alle leverfunksjonstester (som transaminaser, gGT, alkaliske fosfatase, direkte og indirekte bilirubinemi, gallesyrer, blodtelling), dvs. tilstedeværelse av isolert hyperbilirubinemi.
En normal abdominal ultralyd kan også være nyttig og nødvendig for å fastslå normaliteten til leveren og de intra- og ekstrahepatiske galleveiene.
Det er svært vanskelig å utføre en leverbiopsi for en slik diagnose, kanskje bortsett fra når man søker en mer grundig differensialdiagnose i problematiske eller intrikate situasjoner.
Differensialdiagnosen, for å skille Gilbert syndrom fra andre patologier med noen fellestrekk, må fremfor alt gjøres med de andre arvelige patologiene som påvirker bilirubinmetabolismen, nemlig, som nevnt tidligere, Crigler-Najjar I syndrom, som er mye mer alvorlig og har en uheldig prognose; Crigler-Najar II syndrom, mindre bekymringsfullt og med en relativt god prognose, men med høyere indirekte hyperbilirubinemiverdier enn Gilberts og tidligere debut, mest i barndommen, i motsetning til Gilberts, hvor gulsott vanligvis oppstår senere, dvs. rundt 15-18 år av alder.
Rotorsyndrom og Dubin-Johnsons syndrom er også familiære og arvelige hyperbilirubinemier, men med økt direkte eller konjugert bilirubin
De er også patologier av bilirubinmetabolisme, svært sjeldne tilstander, men med forskjellige patogenetiske mekanismer, også med mer intens gulsott; noen ganger kan de også finnes under graviditet eller under østrogenbehandling, noen ganger for å skille fra gulsott av posthepatisk type, men med normale kolestaseindekser.
Det er verdt å nevne andre former for sykelige tilstander preget av økt ukonjugert (eller indirekte) bilirubinemi i blodet som ved Gilberts syndrom: fysiologisk gulsott hos nyfødte og morsmelk gulsott, som oppstår ved fødselen og er godartet og kortvarig av natur. Det er henholdsvis en forsinket modning av bilirubinmetabolismen og, i det andre tilfellet, tilstedeværelsen av forlengelse av fysiologisk gulsott på grunn av en midlertidig endring av glykuronokonjugering ved disposisjon og med genetiske endringer som ligner på Gilberts syndrom.
Andre tilstander med endret bilirubinmetabolisme inkluderer hyperbilirubinemi av legemidler (f.eks. sulfonamid – et bakteriostatisk middel – eller fusidinsyre – et antibiotikum).

Nylig har flere former for leverskade blitt beskrevet hos pasienter med familiær hyperbilirubinukemi, spesielt hos Gilbert; disse pasientene bør overvåkes nøye med tanke på leverfunksjonen når de trenger behandling med en rekke medikamenter, eller ved kjemoterapi.
Til slutt, for fullstendighetens skyld, med hensyn fremfor alt til gulsott, hele det store kapittelet med akutte og kroniske hepatopatier (hepatitt), obstruksjoner av galletreet, og hele kapittelet om kolestase, med hensyn til galle, gallesekresjon, tilstander av hepatocyttinvolvering, og til slutt det store kapittelet hemolyse, der hyperbilirubinemi finnes sammen med endringer i de forskjellige hematokjemiske hematologiske parameterne.
Prognose for Gilbert syndrom
Prognosen for Gilberts syndrom er, som nevnt ovenfor, god og til og med utmerket.
Det er faktisk ingen uønskede effekter på leverfunksjonen eller på hele organismens velvære.
Terapi for Gilberts syndrom
Ingen terapi er nødvendig.
Som et tiltak for å unngå midlertidig økning i hyperbilirubinemi, anbefales det å
- redusere eller forhindre stress
- unngå faste
- ikke misbruk alkohol
- unngå overdreven fysisk anstrengelse.
Les også:
Emergency Live enda mer...Live: Last ned den nye gratisappen til avisen din for iOS og Android
Hepatitt hos barn, her er hva det italienske nasjonale helseinstituttet sier
Gilberts syndrom: Symptomer, årsaker og diagnose av denne leversykdommen
Akutt hepatitt hos barn, Maggiore (Bambino Gesù): 'Gulsott en vekker'
Nobelprisen for medisin til forskere som oppdaget hepatitt C -virus
Leversteatose: hva det er og hvordan du kan forhindre det
Akutt hepatitt og nyreskade på grunn av energidrikkeforbruk: Saksrapport
De forskjellige typene hepatitt: forebygging og behandling
Akutt hepatitt og nyreskade på grunn av energidrikkeforbruk: Saksrapport
Akutt hepatitttilfeller hos barn: lære om viral hepatitt
Hepatisk Steatose: Årsaker og behandling av fettlever
Hepatopati: Ikke-invasive tester for å vurdere leversykdom
Lever: Hva er ikke-alkoholisk Steatohepatitt