
Gaixotasun arraroak: Rothmund-Thomson sindromea
Rothmund-Thomson sindromea larruazaleko gaixotasun arraroa da, 500an Austrian lehen aldiz deskribatu zenetik 1868 pertsona ingurutan deskribatu dena.
Rothmund-Thomson sindromearen agerpenak
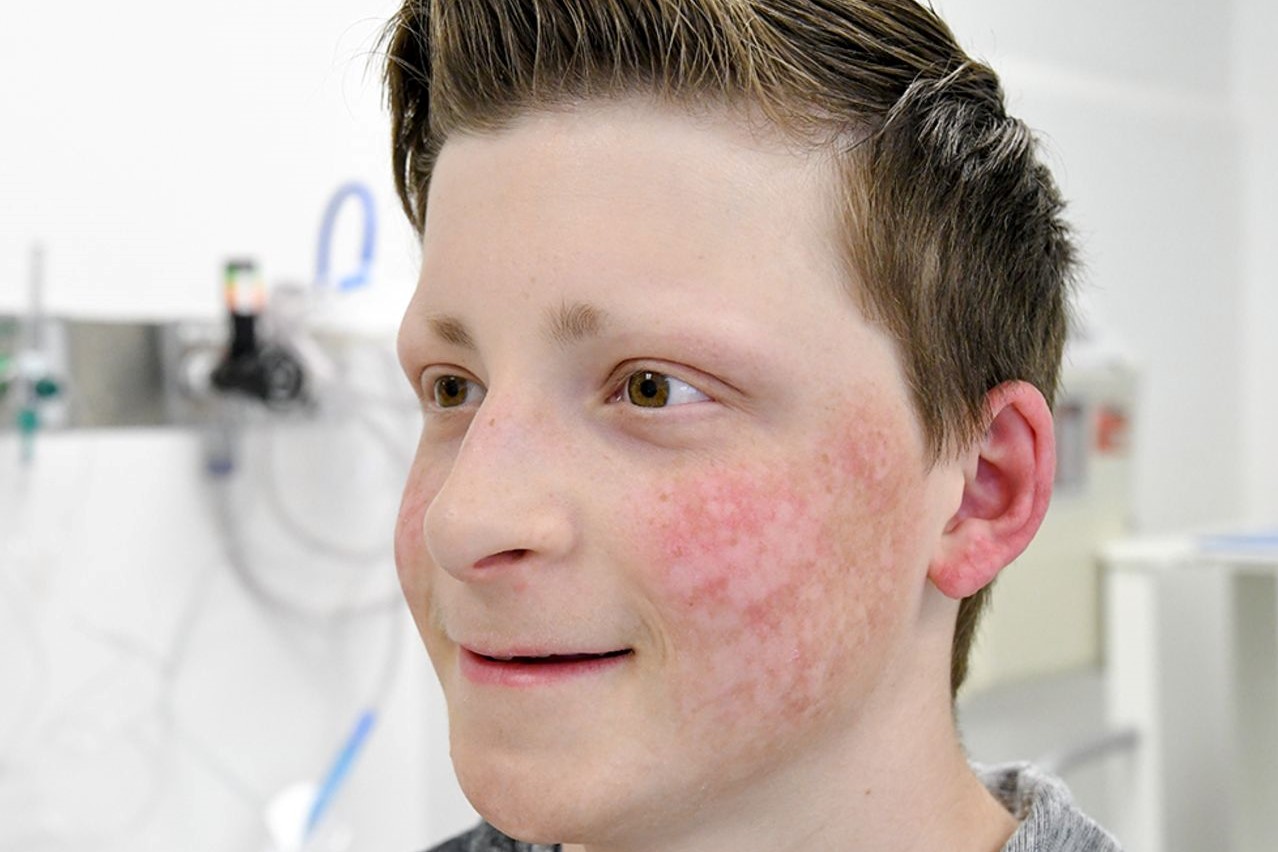
Poikiloderma gisa agertzen da, aurpegiko azaleko erupzio tipikoa, azalaren kanpoaldeko geruzaren (epidermisa) mehetzea (atrofia), azaleko kolorazioaren anomaliak (pigmentazioa) eta azaleko hodien hipo- eta hiperpigmentazioarekin eta dilatazioarekin. azaleko (telangiectasias).
Larruazaleko inplikazioaz gain, larruazal kanpoko sintomak daude, hala nola, jaio aurreko eta ondorengo hazkuntza atzerapena, altuera baxua, betile eta/edo bekain urriak edo eza, hortzetako eta hezurdurako anomaliak, gazteen kataratak, zahartze goiztiarra eta minbizia izateko joera (batez ere osteosarkoma). .
Rothmund-Thomson sindromea kasu gehienetan 4. kromosoman kokatutako RECQL8 helikasa genearen alterazioek (mutazioek) eragindako gaixotasuna da.
Karaktere autosomiko recesibo gisa transmititzen da.
Transmisio autosomiko errezesiboa duten gaixotasunak gene baten bi kopia aldatuta (mutatuta) heredatu dituzten pertsonetan bakarrik gertatzen dira.
Bai amarengandik jasotako kopia eta bai aitarengandik jasotako kopia mutatuta daude.
"Rezesiboa" terminoak esan nahi du bi geneen kopia bakarra aldatzea ez dela nahikoa gaixotasuna eragiteko.
Gaixotasuna eragiteko, geneen bi kopiak mutatu behar dira.
Gurasoak gene alteratuaren kopia bakarraren eramaileak dira (beste kopia normala da), beraz, ez daude gaixorik: eramaile osasuntsuak dira.
Seme-alabak izan nahi dituzten bi eramaile osasuntsuk, haurdunaldi bakoitzean, %25eko aukera dute (lautik bat) haur gaixo bat izateko.
Seme-alaben % 50 eramaile osasuntsuak izango dira (ama eta aita bezala, sintomarik gabe) eta gainerako % 25a osasuntsu egongo da (mutaziorik gabeko genearen bi kopiekin).
Probabilitate hauek jaio gabeko umearen generoaren araberakoak dira.
Orain arte, RECQL4 genearen mutazioak identifikatu dira gutxi gorabehera pazienteen % 60-65etan, eta kaltetutako pertsonen gainerako % 35-40an kausa ezezaguna da.
Rhotmund-Tomson sindromea duten pazienteek erupzio bat izan dezaketen seinale klinikoak dira
Azken hau, normalean, jaiotzean ez dago baina 3 eta 6 hilabete artean garatzen da larruazala gorritu (eritema), hantura eta orbainarekin aurpegian eta gero erupzioa ipurmasailetara eta muturretara zabaltzen da, enborra eta sabelaldea. normalean ez dira kaltetuak.
Erupzioa urteen buruan poikiloderma tipiko bilakatzen da, hiperpigmentazio-eremuak txandakatuz, mehetze-atal txikiak (atrofia) eta hodi berezien dilatazioa (teleangiectasias).
Goiko gorputz-adarretako eskeleto-anomaliak egon daitezke erpuruaren falta edo malformazioarekin edo besaurreak laburtuz.
Beste eskeleto-anomaliak maiz izaten dira, belaunaren eza (aplasia) edo tamainaren murrizketa anormala (hipoplasia) eta osteopenia, hau da, hezurren argaltzea eta ahultzea.
Estatura txikia eta pisu baxua, hortz anomaliak, gastrointestinalaren nahasteak, iltze-displasia, aldebiko kataratak eta osteosarkoma barne dauden tumoreak ere egon daitezke.
Rothmund-Thomson sindromearen diagnostikoa historia klinikoaren bilketa arretatsu batean eta haurraren azterketa berdintsu batean oinarrituta egiten da.
Bereziki poikilodermaren presentzia diagnostikoa bideratzen du
Diagnostikoa larruazaleko aldaketekin lotutako osteosarkoma duten pazienteetan ere kontuan hartu behar da.
Erupzioa atipikoa den kasuetan, diagnostiko klinikoa egin daiteke Rothmund-Thomson sindromearen beste bi ezaugarri daudenean, besteak beste:

- Ile, bekain eta/edo betile urriak eta urriak;
- Estatura laburra pisu baxuaren proportzioan;
- Urdail-hesteetako nahasteak haurtzarotik aurrera agertzen dira, kronikoak adibidez oka eta beherakoa;
- Eskeletiko anomaliak, hala nola erradioaren akatsak, kubitoa, rotularen eza edo hipoplasia, osteopenia;
- Hortzen anomaliak (hortz gutxi garatuak, esmaltearen akatsak edo hortz-hortzetako atzerapena);
- Iltzeen garapen anormala (ilazkal displasikoak);
- oin-zolak loditzea (hiperkeratosia);
- Kataratak, normalean gazteak, aldebikoak;
- Tumoreak, larruazaleko tumoreak (zelula baso-zelulen eta kartzinoma ezkamotsuak) eta osteosarkoma barne.
- Diagnostikoa RECQL4 genearen analisi molekularraren bidez baieztatzen da (gene osoaren sekuentziazioa).
Rothmund-Thomson sindromea diagnostikatzeko irizpide klinikoak daudenean, kasuen % 4-60ean bakarrik identifikatzen dira RECQL65 genearen mutazioak.
Gainerako kasuen %35-40etan, oraindik ezin izan da beste gene arduratsurik identifikatu.
Ez dago tratamendu erabakigarririk.
Rothmund-Thomson sindromea duten haurrak jaiotzatik artatu behar ditu diziplina anitzeko talde batek, eta lehenik eta behin larruazaleko arazoak kudeatzeko eta lesio telangiektasikoen laser tratamendua erabiltzeko espezialistak biltzen dituena.
Taldeak oftalmologo bat ere sartu behar du urtero begi-azterketak egiteko (kataraten presentzia ebaluatzeko eta horien zuzenketa kirurgikoa egiteko).
Irudien ikerketak, hala nola, X izpiak eta CT eskaneatzea ezinbestekoak dira hezur-minbiziarekin lotutako sintomak izanez gero.
Osteopenia eta/edo hausturak dituzten pazienteetan kaltzio eta D bitaminaz osatzea kontuan hartu daiteke.
Pazienteek beroaren eta eguzki-argiaren esposizioa saihestu behar dute, eta horrek erupzioa areagotu dezake pertsona batzuengan eta azaleko minbizia garatzeko arriskua areagotu dezake.
Tumoreak garatzeko arrisku teorikoa kontuan hartuta, hazkunde hormona (GH) ez da gomendatzen GH maila normala duten pertsonentzat, eta GH gabezia duten pazienteentzat, berriz, hazkuntza hormonarekin tratamendu estandarra egokia da.
Rothmund-Thomson sindromearen pronostikoa aldakorra da
Bizi-itxaropena normala da tumorerik ezean, tumoreak dituzten pazienteen pronostikoa, berriz, tumoreen baheketaren eta tratamendu goiztiarren kalitatearen eta maiztasunaren araberakoa da.
Rothmund-Thomson sindromea duten pazienteak bereziki sentikorrak izan daitezke kimioterapiaren albo-ondorioetara eta bigarren mailako tumoreak garatzeko arrisku handia izan dezakete (% 5 larruazaleko minbizia izateko arriskua).
Tratamendua egokia bada, gaixotasunaren forma arinen pronostikoa aldekoa da eta bizi-itxaropena biztanleria orokorraren parekoa da gutxi gorabehera.
Irakurri ere
Gaixotasun ultraarraroak: Malan sindromearen lehen jarraibideak argitaratu dira
Gaixotasun arraroak: Von Hippel-Lindau sindromea
Zika Guillain-Barre sindromearekin lotuta dago ikerketa berri batean
Gaixotasun arraroak: displasia septooptikoa
Gaixotasun arraroak: Sortzetiko hiperinsulinismoa
Oinaren deformazioak: Metatarsus Adductus edo Metatarsus Varus
Progeria: zer da, sintomak, arrazoiak, diagnostikoa eta tratamendu posiblea