
Weichteiltumoren: Leiomyosarkom
Das Leiomyosarkom ist ein Weichteiltumor, der hauptsächlich Frauen und selten Kinder betrifft. Resistenz gegen Chemotherapie, Operation ist die einzige Behandlung
Das Leiomyosarkom ist ein bösartiger Weichteiltumor und gehört zur Familie der Sarkome
Es entwickelt sich aus glatten Muskelzellen.
Es handelt sich um eine Erkrankung des Erwachsenenalters mit einem Häufigkeitsgipfel in der 5. und 6. Lebensdekade und einer Vorliebe für das weibliche Geschlecht, die, wenn auch selten, auch Kinder betreffen kann.
Im pädiatrischen Alter sind Jungen und Mädchen gleichermaßen betroffen.
Leiomyosarkome werden nach der Art der Präsentation in fünf Gruppen eingeteilt:
- „Retroperitoneales Weichteil-Leiomyosarkom“;
- „Leiomyosarkom kutanen Ursprungs“;
- „Leiomyosarkom vaskulären Ursprungs“:
- „Leiomyosarkom bei immungeschwächten Patienten“;
- „Leiomyosarkom des Knochens“.
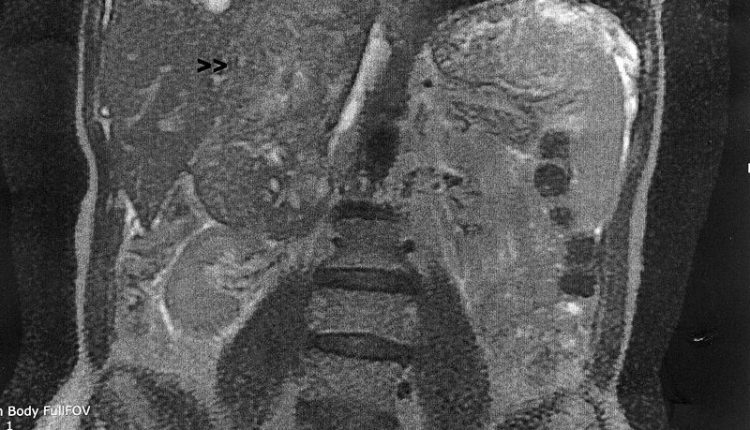
Der häufigste Ort der Entwicklung (50 % der Fälle) ist das Retroperitoneum.
Die Ursache des Auftretens dieses Tumors ist unbekannt und es wurde kein Zusammenhang mit Umweltursachen hergestellt.
Patienten mit hereditärem Retinoblastom, Träger der konstitutionellen Mutation des Rb1-Gens, haben im Vergleich zur Allgemeinbevölkerung ein erhöhtes Risiko, im Erwachsenenalter ein Leiomyosarkom zu entwickeln.
Die Vorliebe für das weibliche Geschlecht hat auch zu Spekulationen geführt, dass Östrogen eine Rolle bei der Proliferation der glatten Muskulatur spielt, jedoch bleibt die Korrelation unklar.
Wie beim Leiomyosarkom bei immungeschwächten Patienten wird normalerweise eine Korrelation mit dem Epstein-Barr-Virus gefunden, und in diesen Fällen kann der Tumor an untypischen Stellen entstehen.
Leiomyosarkome haben, wie alle anderen Sarkome, unspezifische Symptome aufgrund des Vorhandenseins einer Läsion, die umgebende Strukturen komprimiert, disloziert und infiltriert.
Die Darstellungsweise des Leiomyosarkoms variiert daher je nach Ort des Ausbruchs
Das „Weichteil-Leiomyosarkom“ manifestiert sich normalerweise als retroperitoneale Läsion, in diesem Fall sind die Symptome vage abdominale Beschwerden, die mehr oder weniger mit Gewichtsverlust einhergehen.
Das Leiomyosarkom kutanen Ursprungs zeigt sich normalerweise als kleine intradermale Schwellung.
Das Leiomyosarkom vaskulären Ursprungs entwickelt sich aus großen Blutgefäßen und befällt häufig die Pulmonalarterie.
In diesem Fall sind die Symptome hauptsächlich Atembeschwerden und Brustschmerzen aufgrund einer Behinderung des Blutflusses.
Das Leiomyosarkom der Knochen ist eine sehr seltene Entität und betrifft normalerweise die langen Knochen
Bei Verdacht auf das Vorhandensein eines Sarkoms muss aufgrund der objektiven Untersuchung und der radiologischen Voruntersuchung ein Fragment des Tumors entnommen und vom Pathologen (möglicherweise ein Experte für diese Art von Läsion) analysiert werden, der Facharzt kann dies vornehmen die Diagnose.
Es gibt auch weitere Untersuchungen, die die Eigenschaften und das Ausmaß dieses Tumors beurteilen:
- Kernspinresonanztomographie (MRT) und/oder Computertomographie (CT), die es ermöglichen, die Größe des Tumors, seine Ausdehnung und Beziehung zu benachbarten Strukturen zu beurteilen;
- Röntgen- oder CT-Scan des Brustkorbs, Plus- oder Minus-Knochenszintigraphie zur Beurteilung entfernter sekundärer Läsionen.
- Eine Operation ist die Hauptstütze der Behandlung des Leiomyosarkoms und kann in Fällen, in denen die Läsion radikal entfernt wird, die einzige Behandlung sein.
Bei unvollständiger Exzision kann eine ergänzende lokale Strahlentherapie erforderlich sein, insbesondere bei Läsionen der Extremitäten.
Das Leiomyosarkom ist kein Chemotherapie-empfindlicher Tumor

Die Rolle der zytostatischen Behandlung ist unklar und folglich wird die Chemotherapie ausgewählten Fällen vorbehalten: große Läsionen, die zunächst inoperabel sind, mit dem Ziel, das Tumorvolumen und die damit verbundenen Symptome zu reduzieren; Patienten mit Läsionen, die aus der Ferne wiederkehren.
Die am häufigsten verwendeten Chemotherapeutika sind Anthrazykline, Ifosfamid, Gemcitabin, Taxotere, Dacarbazin und Trabectidin.
In Bezug auf Therapien, die auf die Tumorzelle abzielen, ist von den verschiedenen getesteten Molekülen das einzige, das eine mäßige Wirksamkeit gegen Leiomyosarkom gezeigt hat, Pazopanib, ein Tyrosinkinase-Hemmer, der oral verabreicht wird.
Die Prognose eines Patienten mit Leiomyosarkom hängt von den histologischen Merkmalen der Läsion, der lokalen Ausdehnung (Größe der Raumforderung und Beziehung zu umgebenden Strukturen) und der anatomischen Lokalisation der Läsion sowie einem entfernten Rezidiv ab.
Wenn die gesamte Tumormasse entfernt werden kann, ist die Prognose mit einer Heilungschance von 80 % sehr gut.
Extremitätentumoren haben eine bessere Prognose als retroperitoneale Tumoren, da es sich um kleinere Läsionen handelt und die Chance einer radikalen chirurgischen Entfernung höher ist.
Intradermale Läsionen haben eine ausgezeichnete Prognose, da sie dazu neigen, lokalisiert zu bleiben und nicht zu metastasieren.
Die Prognose bleibt weniger günstig für Patienten mit entfernten rezidivierenden Läsionen.
Lesen Sie auch
Weichteilsarkome: Bösartiges fibröses Histiozytom
Hirntumoren: Symptome, Klassifizierung, Diagnose und Behandlung
Pädiatrische Hirntumoren: Arten, Ursachen, Diagnose und Behandlung
Hirntumore: CAR-T bietet neue Hoffnung für die Behandlung von inoperablen Gliomen
Lymphom: 10 nicht zu unterschätzende Alarmglocken
Chemotherapie: Was es ist und wann es durchgeführt wird
CAR-T: Eine innovative Therapie für Lymphome
Was ist CAR-T und wie funktioniert CAR-T?
Strahlentherapie: Wofür sie verwendet wird und was die Auswirkungen sind
Kraniosynostose-Chirurgie: Übersicht
Pädiatrische Malignome: Medulloblastom