
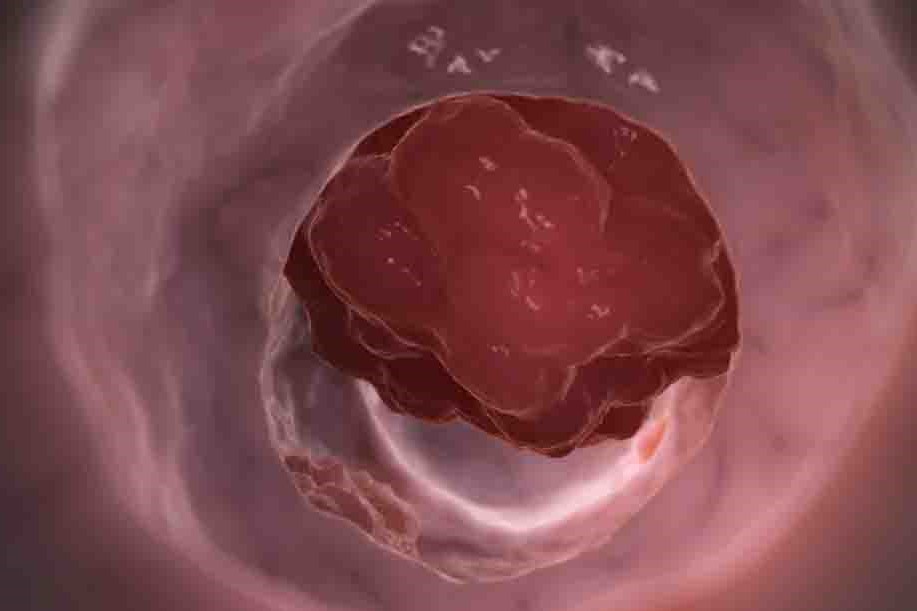
Gastroenterology: intestinal polyps and polyposis in paediatrics
Intestinal polyps are protuberances of the tissues of the intestinal wall that protrude into the bowel cavity
Polyps are a rare occurrence in children
There are two distinct conditions:
- The single isolated polyp;
- The intestinal polyposis.
The single isolated polyp of the rectum/intestine, which is almost always benign in nature and without risk of possible malignant degeneration (juvenile polyp), mostly manifests itself with episodes of rectal bleeding (bright red blood and mucus with the stools).
As a result of the bleeding, a quarter to a third of children with a single isolated polyp experience iron deficiency anaemia.
It is diagnosed by endoscopic test (recto-colonoscopy).
Treatment consists of endoscopically removing (resecting) the polyp under deep sedation.
This type of polyp does not require further examination or control, except in the event of new rectal bleeding.
They are characterised by the presence of numerous polyps that can undergo malignant transformation and have a genetic cause.
The most frequent intestinal polyposes are:
- Familial Adenomatous Polyposis (PAF);
- Hamartomatous polyposis, the most common of which is Peutz Jeghers Syndrome;
- Juvenile Polyposis Syndromes.
Familial Adenomatous Polyposis
Familial Adenomatous Polyposis (PAF) is a rare genetic syndrome, with an incidence of 1 in 8000 individuals, characterised by the appearance, usually as early as pre-adolescence/adolescent age (8-12 years), of hundreds or thousands of polyps (adenomas) in the colon and rectum.
If left untreated, Familial Adenomatous Polyposis (PAF) progresses to the development of colorectal cancer which usually occurs before the age of 40, more rarely in adolescence.
Patients may also develop various extra-intestinal manifestations that include desmoid tumours (10-30%), osteomas of the skull or jaw, sebaceous cysts, ocular defects (hypertrophy of the pigmented epithelium of the retina) but also adrenal adenoma (7-13%), cancers of the duodenum (5-11%), pancreas (2%), thyroid (2%), brain (medulloblastomas over 1%) and liver (hepatoblastoma of children over 5 years in 0.7%).
A less aggressive variant is attenuated familial adenomatous polyposis, characterised by a smaller number of colo-rectal adenomatous polyps (usually between 10 and 100), predominantly localised in the right colon, with the appearance of adenomas at a later age and low risk of cancer.
Familial Adenomatous Polyposis is a hereditary disease, caused by mutations in the APC (Adenomatous Polyposis Coli) gene, which are transmitted from parents to offspring in an autosomal dominant manner, i.e. the affected parent has a 50% chance of transmitting the disease to each of his or her offspring, regardless of the sex of the unborn child.
In 15-20% of cases, mutations are ‘de novo’, i.e. newly arising, and are therefore not inherited from the parents but occur during the formation of the egg or sperm cell or in the very early stages of embryonic development.
In such a case, no other family member will be ill apart from the person carrying the genetic defect.
Numerous mutations (approximately 400) have already been described and are responsible for the different clinical course and manifestations found among patients, even within the same family.
Subjects are often asymptomatic or may present with blood in the stool, abdominal pain, and progressive anaemia.
Diagnosis is based on endoscopic examination (detection of more than 100 adenomatous polyps at colonoscopy) and/or genetic testing (search for the APC gene mutation by taking blood).
If the APC gene mutation is confirmed, the genetic test must be extended to all first-degree relatives.
Once a diagnosis has been made, it is essential to carry out periodic surveillance to prevent the development of both intestinal and extra-intestinal problems.
The timing of screening and endoscopic surveillance has been defined by the European Society for Paediatric Gastroenterology Hepatology and Nutrition (ESPGHAN) taking into account the specific risk of neoplastic transformation of intestinal polypoid lesions (colorectal carcinoma, gastric and duodenal carcinoma), so it is recommended that the first endoscopic checks should begin from the age of 12 in the absence of symptoms.
Children of parents with familial adenomatous polyposis can be evaluated for hepatoblastoma from birth to 5 years of age by measuring serum alpha-fetoprotein levels and possibly by liver ultrasound;
thyroid ultrasound should be performed from adolescence and repeated every 3-5 years. An annual clinical evaluation is indicated for the prevention of medulloblastoma as well as desmoids.
Treatment of familial adenomatous polyposis (PAF) involves removal of the colon (prophylactic total colectomy) to prevent progression to cancer.
The surgical procedure is planned according to time (pre-adolescence/adult/adult age) and modality (laparoscopic technique) to be established according to each patient’s clinical course (number and size of polypoid lesions, degree of dysplasia), as well as the psycho-social needs of the patient and family.
The surgical technique (total colectomy with or without removal of the rectum) can be defined in accordance with the characteristics of each individual subject (e.g. number of polyps in the rectum, predisposition to the development of desmoids according to the type of genetic mutation, etc.), also sharing the possible early and late risks, which may have an impact on quality of life.
In fact, total colectomy with ileo-rectum anastomosis entails the preservation of the rectum, which, if on the one hand favours a good control of evacuations, on the other hand implies periodic endoscopic controls, even every 3-6 months, for the remediation (removal of polyps by endoscopy) of this residual tract; total procto-colectomy with ileo-anastomosis on J pouch ileum is instead a more radical procedure, where the rectum is also removed, but characterised by a higher number of daily evacuations.
The paediatric patient with Familial Adenomatous Polyposis (PAF) must, on reaching the age of majority, continue endoscopic and ultrasonographic controls at the adult reference centres following a transition path involving the sending paediatric centre and the receiving adult centre.
Peutz Jeghers syndrome
Peutz Jeghers Syndrome (SPJ) is a genetic disorder caused by an alteration (mutation) in the STK11/LKB1 gene.
It is a rare disease that affects one in 75,000 to 300,000 newborns.

It is characterised by the presence of many benign and usually non-degenerating polyps scattered throughout the gastrointestinal tract, associated in most cases with lentiginous ‘spots’ on the mucous membranes and skin (lips and mouth, palms of the hands, soles of the feet, perianal and genital region).
These ‘spots’ appear early in life and, although those on the skin may disappear, those on the mouth remain and are very useful for diagnosis.
The STK11/LKB1 gene mutation is inherited in an autosomal dominant manner.
However, about half of the patients have no family member with Peutz Jeghers syndrome.
These are the ‘de novo’ mutations mentioned above.
The diagnosis is based on clinical criteria (presence of freckled spots), genetic tests (STK11 gene mutation), episodes of bright red blood in the stool (rectorrhagia), abdominal pain, episodes of intestinal invagination and the presence of polyps, even large ones, in the stomach, duodenum, colon and small intestine (jejunum and ileum); In the latter case, the presence of a large polyp occupying the entire intestinal cavity can lead to intestinal obstruction with a picture of an ‘acute abdomen’ requiring surgery.
Observed under the microscope, polyps are hamartomatous in nature
Hamartomas are benign, tumour-like neoformations composed of a variety of cell types that grow in a disorderly fashion.
The most frequent sites are:
- The small intestine (60-90%);
- The colon (50-60%);
- The stomach (49%);
- The rectum (32%).
The instrumental examinations for diagnosis and the surveillance programme (polyp sampling, histological test and remediation) are represented by:
- Gastroscopy (for the study of the oesophagus, stomach and duodenum);
- Colonoscopy (for the study of the colon);
- Videocapsule (for the study of the small intestine);
- Single or double-balloon entheoscopy (for the study of the small intestine);
- Complete abdominal ultrasound;
- Thyroid ultrasound;
- Testicular ultrasound.
The syndrome can have intestinal and extra-intestinal complications and in particular tumours:
- Of the colon (39%), pancreas (36%), stomach (29%) and small intestine (13%), lung, ovarian, testicular and breast.
- The surveillance programme of patients diagnosed with Peutz Jeghers syndrome should start from the age of 8 years if the patient is asymptomatic (absence of clinical symptoms), before the age of 8 years if symptoms are present.
Juvenile polyposis syndrome (JPS)
Juvenile polyposis syndrome is a rare, autosomal dominant condition characterised by the presence of multiple hamartomatous polyps (greater than 5) distributed along the gastrointestinal tract.
It may be associated with a high risk of cancerous lesions of the digestive tract starting mainly from the age of 18 years (rare before the age of 18 years).
Juvenile polyposis syndromes (PJS) may present clinically with rectorrhagia (blood in the stool), anaemia, abdominal pain and hypoalbuminaemia and are genetically diagnosable by genetic testing with mutations present in 60% of cases.
Although similar to Peutz Jeghers syndrome, it is distinguished by phenotypic variables associated with PTEN (Hamartoma tumour syndrome PHTS) mutation (SMAD 4-BMPR1A)
These include:
- Cowden syndrome (intestinal polyps, macrocephaly, mental retardation);
- Ruvalcaba syndrome.
The surveillance programme involves endoscopic assessment (gastroscopy, colonoscopy + video capsule) depending on the clinical symptoms, the number of polyps and the histological nature.
Endoscopic screening tends to be performed from the age of 12 in children with a family history and who are symptomatic.
An imaging study (brain and cardiac MRI) is important due to the high risk of cerebral and large vessel arteriovenous malformations that may cause severe haemorrhage.
Read Also
Emergency Live Even More…Live: Download The New Free App Of Your Newspaper For IOS And Android
Endoscopic Polypectomy: What It Is, When It Is Performed
Juvenile Gastrointestinal Polyposis: Causes, Symptoms, Diagnosis, Therapy
Intestinal Polyps: Diagnosis And Types
Differences Between Mechanical And Paralytic Ileus: Causes, Symptoms And Treatment
Short Bowel Syndrome: Causes, Therapy, Diet
Vomiting Blood: Haemorrhaging Of The Upper Gastrointestinal Tract
Pinworms Infestation: How To Treat A Paediatric Patient With Enterobiasis (Oxyuriasis)
Intestinal Infections: How Is Dientamoeba Fragilis Infection Contracted?
Gastrointestinal Disorders Caused By NSAIDs: What They Are, What Problems They Cause