
Rare Diseases: Rothmund-Thomson Syndrome
Rothmund-Thomson syndrome is a very rare skin disease that has been described in around 500 individuals since it was first described in Austria in 1868
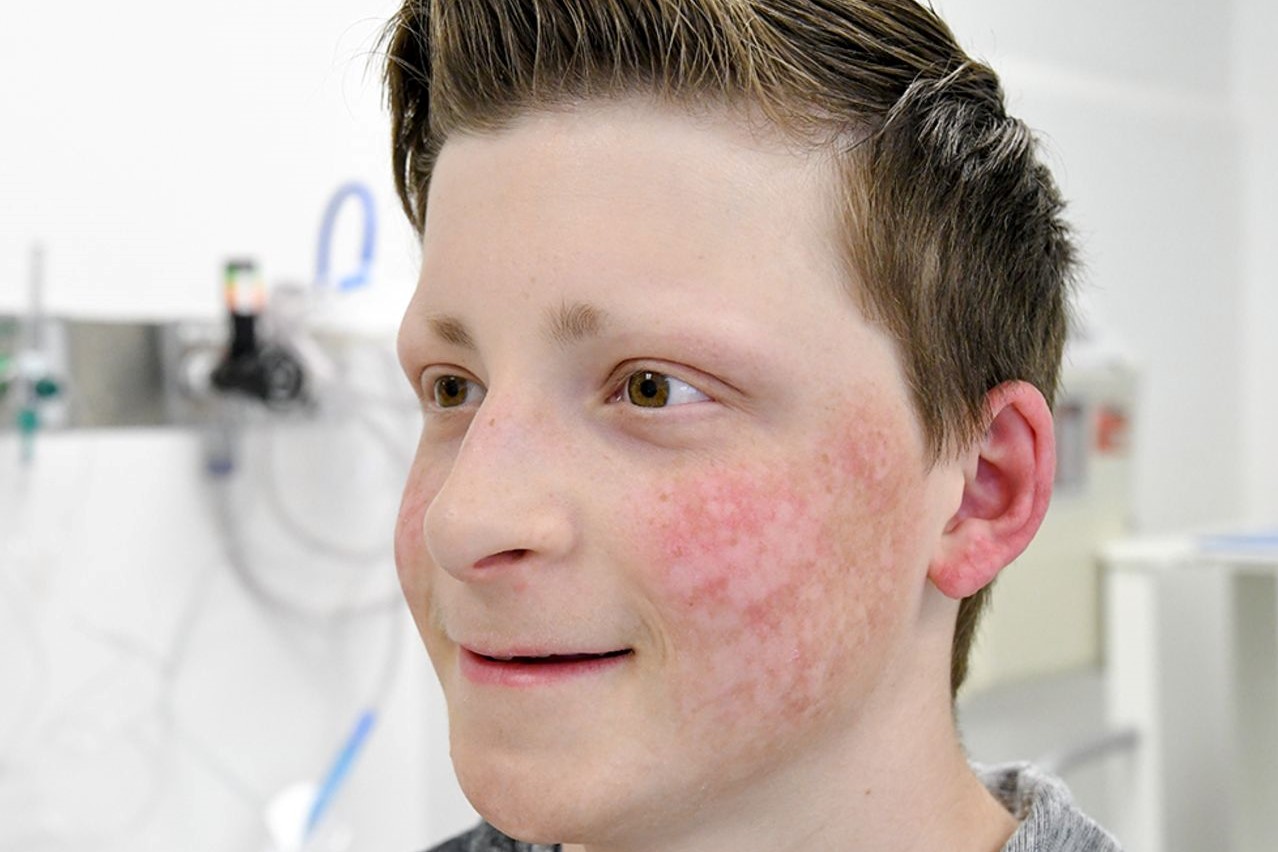
Manifestations of Rothmund-Thomson syndrome
It manifests as poikiloderma, a typical facial skin rash characterised by thinning (atrophy) of the outermost layer of the skin (the epidermis), abnormalities in the colouring (pigmentation) of the skin with both hypo- and hyperpigmentation and dilation of the superficial vessels of the skin (telangiectasias).
In addition to skin involvement, there are extracutaneous symptoms such as both pre- and postnatal growth retardation, short stature, sparse or absent eyelashes and/or eyebrows, dental and skeletal abnormalities, juvenile cataracts, premature ageing and predisposition to cancer (especially osteosarcoma).
Rothmund-Thomson syndrome is a disease caused in most cases by alterations (mutations) in the RECQL4 helicase gene located on chromosome 8
It is transmitted as an autosomal recessive character.
Diseases with autosomal recessive transmission only occur in people who have inherited two altered (mutated) copies of a gene.
Both the copy inherited from the mother and the copy inherited from the father are mutated.
The term ‘recessive’ means that the alteration of only one of the two gene copies is not sufficient to cause the disease.
To cause the disease, both copies of the genes must be mutated.
The parents are carriers of only one copy of the altered gene (the other copy is normal) so they are not sick: they are healthy carriers.
Two healthy carriers who want to have children have, at each pregnancy, a 25% chance (one in four) of having a sick child.
50% of their children will be healthy carriers (like mother and father, without symptoms) and the remaining 25% will be healthy (with both copies of the gene without the mutation).
These odds are independent of the gender of the unborn child.
To date, mutations in the RECQL4 gene have been identified in approximately 60-65% of patients, while in the remaining 35-40% of affected individuals the cause is unknown.
Clinical signs that patients with Rhotmund-Tomson syndrome may present with include a rash
The latter is usually not present at birth but develops between 3 and 6 months of age with a reddening of the skin (erythema), swelling and scarring on the face and subsequent spread of the rash to the buttocks and extremities, while the trunk and abdomen are usually not affected.
The rash evolves after years into the typical poikiloderma with alternating areas of hyperpigmentation, small patches of thinning (atrophy) and dilatation of supeficial vessels (teleangiectasias).
Skeletal abnormalities of the upper limbs may be present with absence or malformation of the thumb or shortening of the forearms.
Other frequent skeletal abnormalities are the absence (aplasia) or abnormal reduction in size (hypoplasia) of the kneecap and osteopenia, i.e. bone thinning and weakening.
Short stature and low body weight, dental abnormalities, gastrointestinal disorders, nail dysplasia, bilateral cataracts, and tumours including osteosarcoma may also be present.
The diagnosis of Rothmund-Thomson syndrome is made on the basis of a careful collection of the clinical history and an equally careful examination of the child.
The presence of poikiloderma in particular points towards the diagnosis
The diagnosis must also be considered in patients with osteosarcoma associated with skin changes.
In cases where the rash is atypical, the clinical diagnosis can be made when at least two other features of Rothmund-Thomson syndrome are present, including:

- Sparse and sparse hair, eyebrows and/or eyelashes;
- Short stature in proportion to low weight;
- Gastrointestinal disorders present from childhood onwards, such as chronic vomiting and diarrhoea;
- Skeletal abnormalities such as defects of the radius, ulna, absence or hypoplasia of the patella, osteopenia;
- Tooth abnormalities (underdeveloped teeth, enamel defects or delayed teething);
- Abnormal nail development (dysplastic nails);
- Thickening (hyperkeratosis) of the soles of the feet;
- Cataracts, usually juvenile, bilateral;
- Tumours, including skin tumours (basal cell and squamous cell carcinoma) and osteosarcoma.
- The diagnosis is then confirmed by molecular analysis of the RECQL4 gene (whole gene sequencing).
When the clinical criteria for diagnosing Rothmund-Thomson syndrome are present, mutations in the RECQL4 gene are identified in only 60-65% of cases
In the remaining 35-40% of cases, it has not yet been possible to identify another responsible gene.
There is no decisive treatment.
Children with Rothmund-Thomson syndrome must be cared for from birth by a multidisciplinary team that includes first and foremost specialists for the management of skin problems and the use of laser treatment of telangiectasic lesions.
The team must also include an ophthalmologist to perform annual eye examinations (to assess the presence of cataracts and for their possible surgical correction).
Imaging investigations such as X-rays and CT scans are essential in case of symptoms potentially associated with bone cancer.
Supplementation with calcium and vitamin D may be considered in patients with osteopenia and/or fractures.
Patients should avoid exposure to heat and sunlight, which may aggravate the rash in some individuals and increase the risk of developing skin cancer.
Given the theoretical risk of developing tumours, growth hormone (GH) is not recommended for individuals with normal GH levels, while for patients with GH deficiency, standard treatment with growth hormone is appropriate.
The prognosis of Rothmund-Thomson syndrome is variable
Life expectancy is normal in the absence of tumours, while the prognosis of patients with tumours depends on the quality and frequency of tumour screening and early treatment.
Patients with Rothmund-Thomson syndrome may be particularly sensitive to the side effects of chemotherapy and have a high risk of developing secondary tumours (5% risk of developing skin cancer).
If treatment is adequate, the prognosis of mild forms of the disease is favourable and life expectancy is approximately comparable to that of the general population.
Read Also
Emergency Live Even More…Live: Download The New Free App Of Your Newspaper For IOS And Android
Rare Diseases: Russian Economist Anatoly Chubais Diagnosed With Guillain Barré Syndrome
Ultrarare Diseases: First Guidelines For Malan Syndrome Published
Rare Diseases: Von Hippel-Lindau Syndrome
Zika Linked To Guillain-Barre Syndrome In New Study
Rare Diseases: Septo-Optic Dysplasia
Rare Diseases: Congenital Hyperinsulinism
Foot Deformities: Metatarsus Adductus Or Metatarsus Varus
Progeria: What Is It, Symptoms, Causes, Diagnosis And Possible Treatment